

Identification of GAA variants through whole exome sequencing targeted to a cohort of 606 patients with unexplained limb-girdle muscle weakness
- PMID: 29149851
- PMCID: PMC5693551
- DOI: 10.1186/s13023-017-0722-1
Identification of GAA variants through whole exome sequencing targeted to a cohort of 606 patients with unexplained limb-girdle muscle weakness
Abstract
Background: Late-onset Pompe disease is a rare genetic neuromuscular disorder caused by a primary deficiency of α-glucosidase and the associated accumulation of glycogen in lysosomal vacuoles. The deficiency of α-glucosidase can often be detected using an inexpensive and readily accessible dried blood spot test when Pompe disease is suspected. Like several neuromuscular disorders, Pompe disease typically presents with progressive weakness of limb-girdle muscles and respiratory insufficiency. Due to the phenotypic heterogeneity of these disorders, however, it is often difficult for clinicians to reach a diagnosis for patients with Pompe disease. Six hundred and six patients from a European population were recruited onto our study. Inclusion criteria stipulated that index cases must present with limb-girdle weakness or elevated serum creatine kinase activity. Whole exome sequencing with at least 250 ng DNA was completed using an Illumina exome capture and a 38 Mb baited target. A panel of 169 candidate genes for limb-girdle weakness was analysed for disease-causing variants.
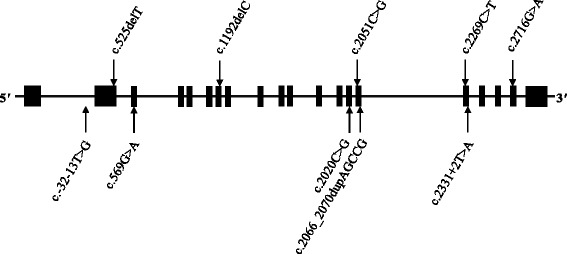
Results: A total of 35 variants within GAA were detected. Ten distinct variants in eight unrelated index cases (and four siblings not sequenced in our study) were considered disease-causing, with the patients presenting with heterogeneous phenotypes. The eight unrelated individuals were compound heterozygotes for two variants. Six patients carried the intronic splice site c.-13 T > G transversion and two of the six patients also carried the exonic p.Glu176ArgfsTer45 frameshift. Four of the ten variants were novel in their association with Pompe disease.
Conclusions: Here, we highlight the advantage of using whole exome sequencing as a tool for detecting, diagnosing and treating patients with rare, clinically variable genetic disorders.
Keywords: Pompe disease; Sequence variants; Whole exome sequencing.
Conflict of interest statement
Ethics approval and consent to participate
Full ethics approval was granted for the Newcastle MRC Centre Biobank for Rare and Neuromuscular Diseases by the Newcastle and North Tyneside research ethics committee (REC reference number 09/H0906/28). All patients provided informed consent for the storage and use of their biomaterial in the Newcastle MRC Centre Biobank for Rare and Neuromuscular Diseases.
Consent for publication
Not applicable.
Competing interests
VS is or has been a principal investigator for trials sponsored by Sanofi Genzyme, GSK, Prosensa/BioMarin Pharmaceuticals, Ionis Pharmceuticals and Sarepta Therapeutics. VS received speaker honoraria from Sanofi Genzyme. For the last 3 years VS is or has been on advisory boards for Audentes Therapeutics, Biogen, Bristol-Myer Squibb, Italfarmaco S.p.A., Sarepta Therapeutics, Summit Therapeutics, Tivorsan Pharmaceuticals, TrophyNOD, and Wave Therapeutics. KGC is principal investigator for trials sponsored by Sanofi Genzyme and KGC received speaker honoraria and travel grants from Sanofi Genzyme. The authors’ declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Screening for Pompe disease in Serbian patients with limb-girdle muscle weakness.Clin Neurol Neurosurg. 2025 Jul;254:108950. doi: 10.1016/j.clineuro.2025.108950. Epub 2025 May 7. Clin Neurol Neurosurg. 2025. PMID: 40359611
-
The frequency of late-onset Pompe disease in pediatric patients with limb-girdle muscle weakness and nonspecific hyperCKemia: A multicenter study.Neuromuscul Disord. 2016 Nov;26(11):796-800. doi: 10.1016/j.nmd.2016.09.001. Epub 2016 Sep 6. Neuromuscul Disord. 2016. PMID: 27666774
-
Detection of variants in dystroglycanopathy-associated genes through the application of targeted whole-exome sequencing analysis to a large cohort of patients with unexplained limb-girdle muscle weakness.Skelet Muscle. 2018 Jul 30;8(1):23. doi: 10.1186/s13395-018-0170-1. Skelet Muscle. 2018. PMID: 30060766 Free PMC article.
-
Newborn screening for Pompe disease in Japan: report and literature review of mutations in the GAA gene in Japanese and Asian patients.J Hum Genet. 2019 Aug;64(8):741-755. doi: 10.1038/s10038-019-0603-7. Epub 2019 May 10. J Hum Genet. 2019. PMID: 31076647 Review.
-
Pompe disease: pathogenesis, molecular genetics and diagnosis.Aging (Albany NY). 2020 Aug 3;12(15):15856-15874. doi: 10.18632/aging.103794. Epub 2020 Aug 3. Aging (Albany NY). 2020. PMID: 32745073 Free PMC article. Review.
Cited by
-
Navigating Pompe Disease Assessment: A Comprehensive Scoping Review.Cureus. 2024 Nov 13;16(11):e73593. doi: 10.7759/cureus.73593. eCollection 2024 Nov. Cureus. 2024. PMID: 39677172 Free PMC article.
-
The clinical-phenotype continuum in DYNC1H1-related disorders-genomic profiling and proposal for a novel classification.J Hum Genet. 2020 Nov;65(11):1003-1017. doi: 10.1038/s10038-020-0803-1. Epub 2020 Aug 12. J Hum Genet. 2020. PMID: 32788638 Free PMC article.
-
The Latin American experience with a next generation sequencing genetic panel for recessive limb-girdle muscular weakness and Pompe disease.Orphanet J Rare Dis. 2020 Jan 13;15(1):11. doi: 10.1186/s13023-019-1291-2. Orphanet J Rare Dis. 2020. PMID: 31931849 Free PMC article.
-
Genetic cause of heterogeneous inherited myopathies in a cohort of Greek patients.Mol Genet Metab Rep. 2020 Nov 30;25:100682. doi: 10.1016/j.ymgmr.2020.100682. eCollection 2020 Dec. Mol Genet Metab Rep. 2020. PMID: 33304817 Free PMC article.
-
A novel compound heterozygous mutation in the POMK gene causing limb-girdle muscular dystrophy-dystroglycanopathy in a sib pair.Neuromuscul Disord. 2018 Jul;28(7):614-618. doi: 10.1016/j.nmd.2018.04.012. Epub 2018 May 16. Neuromuscul Disord. 2018. PMID: 29910097 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
