

Lysosomal storage diseases
- PMID: 29152458
- PMCID: PMC5685203
- DOI: 10.3233/TRD-160005
Lysosomal storage diseases
Abstract
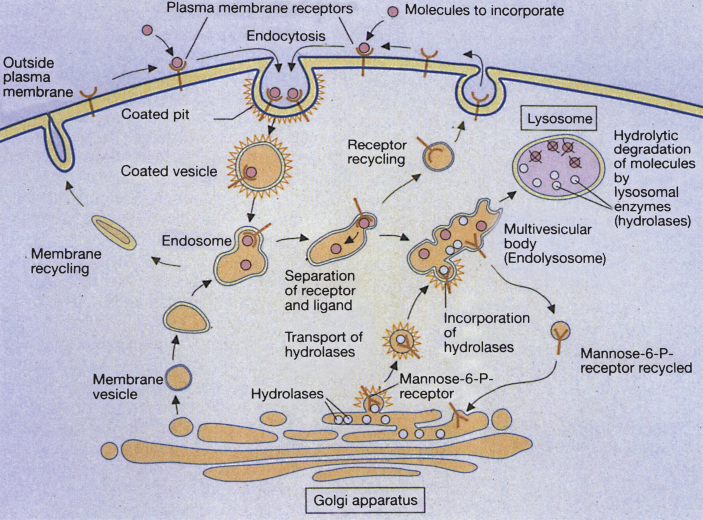
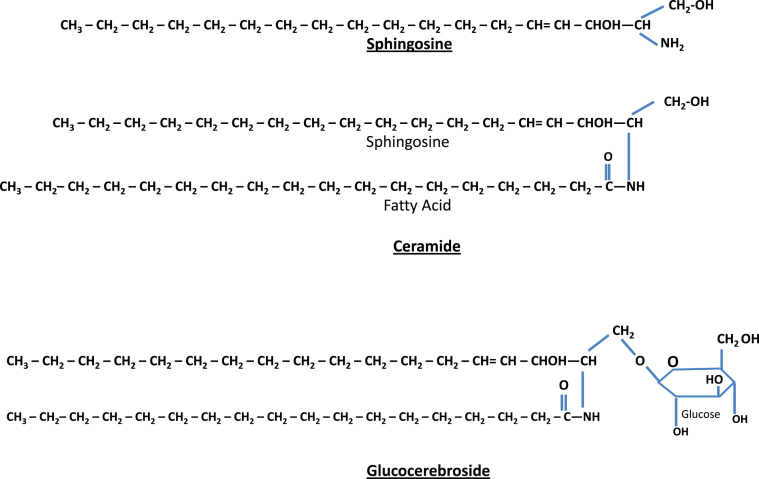
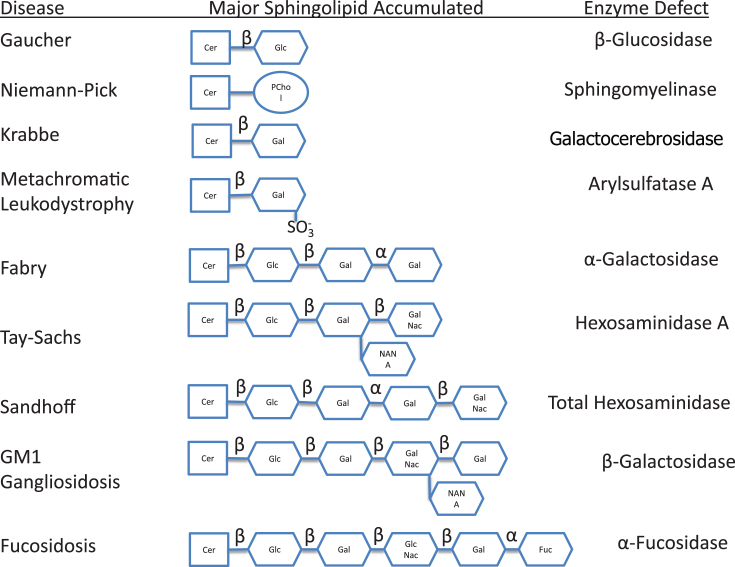
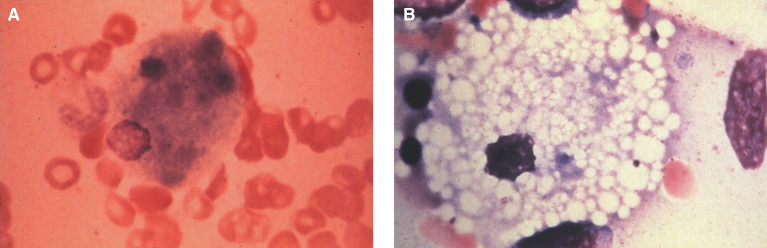
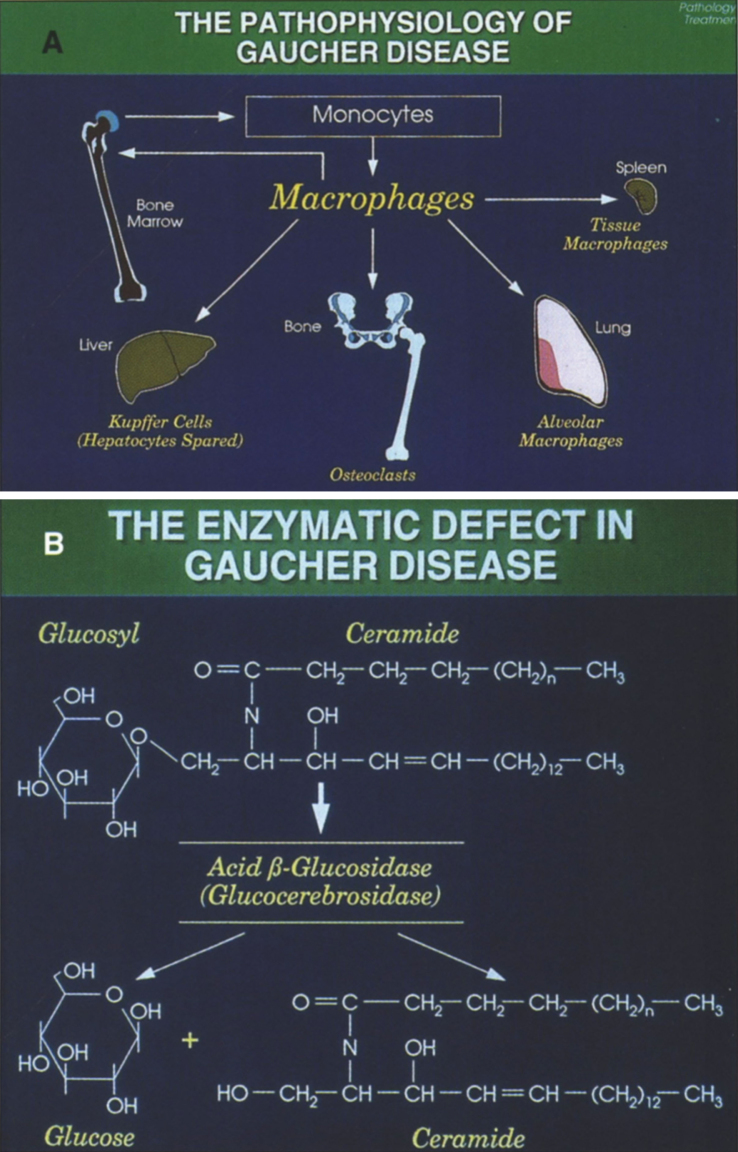
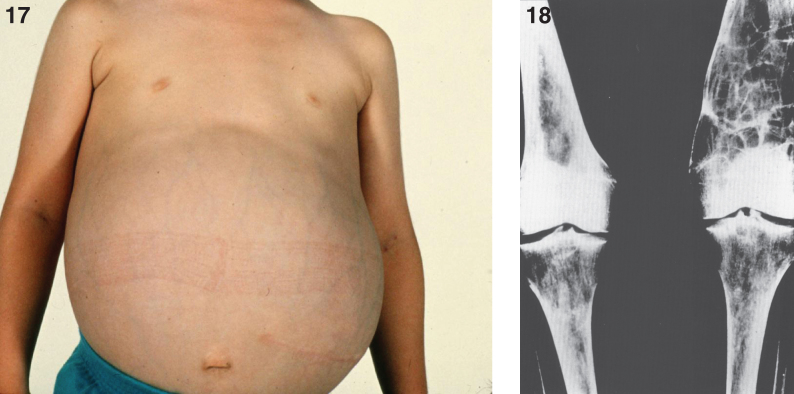
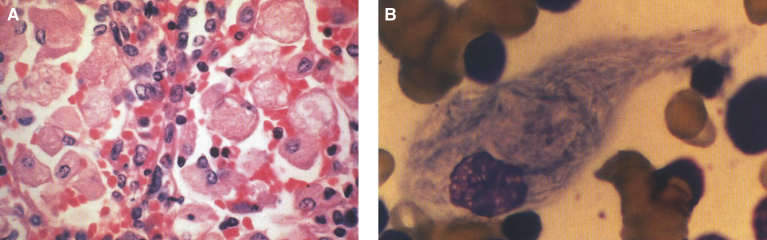
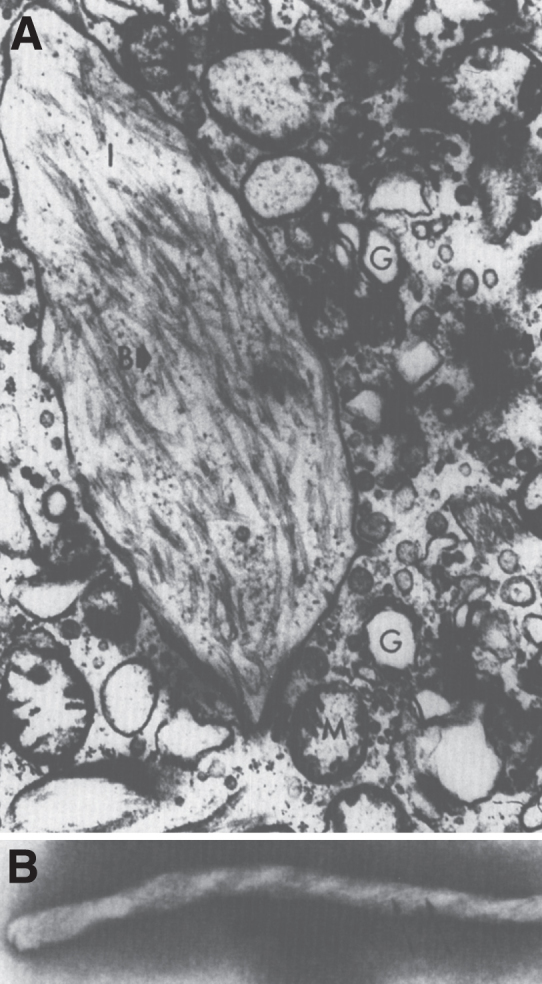
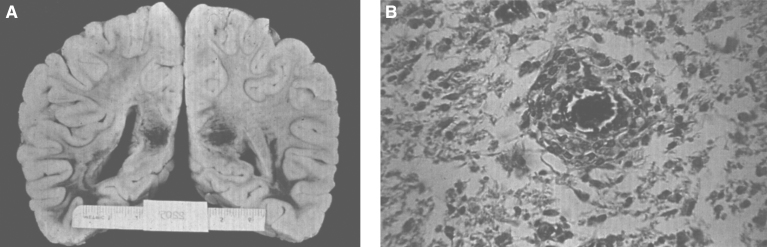
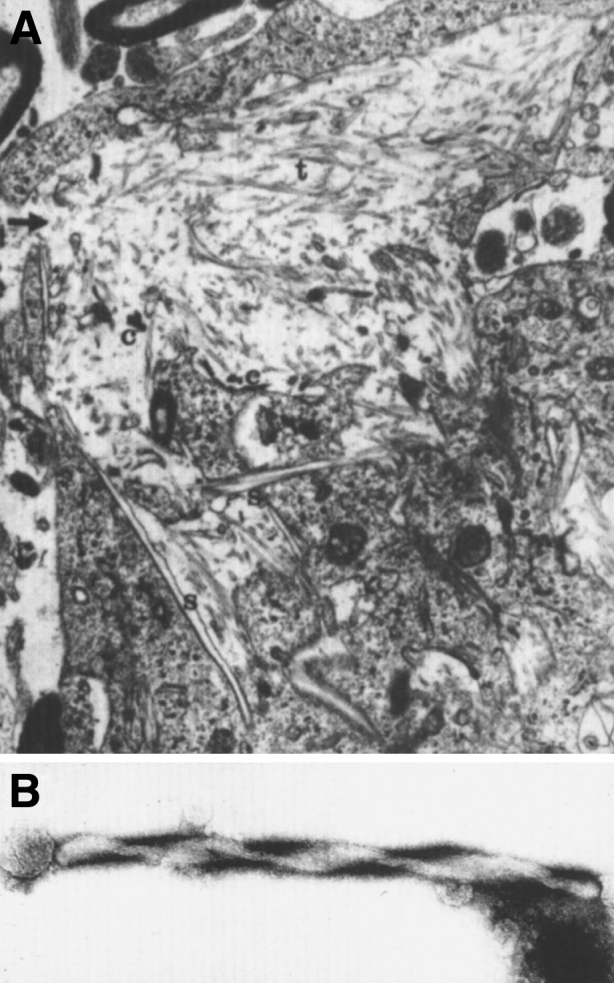
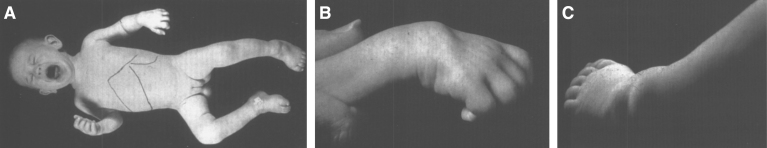
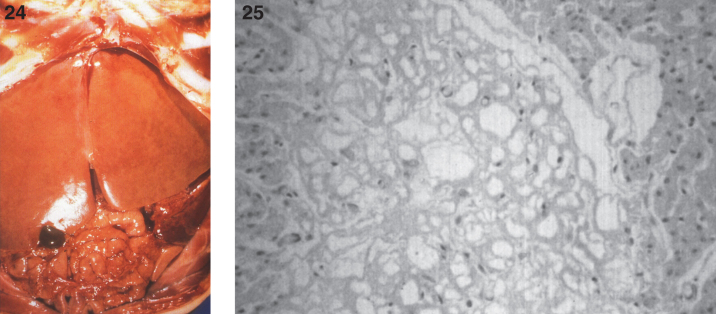
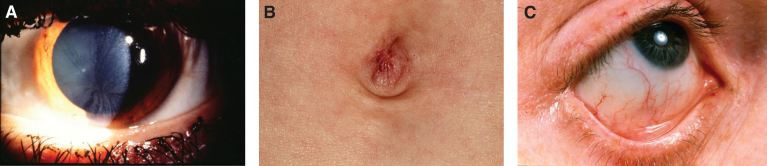
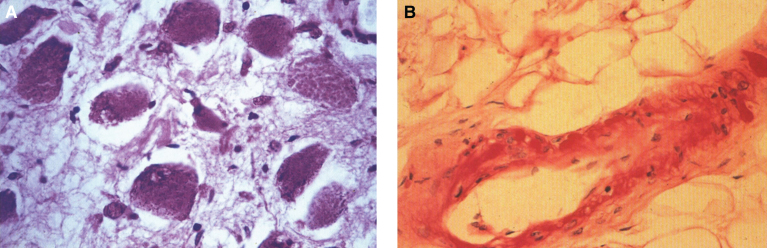
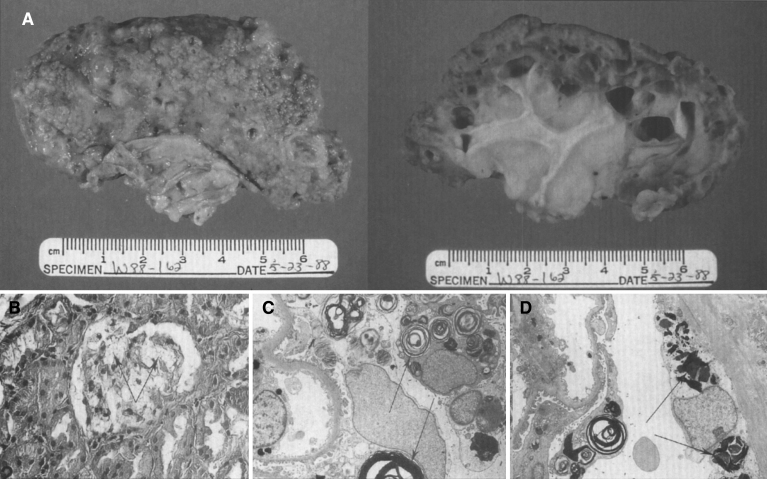
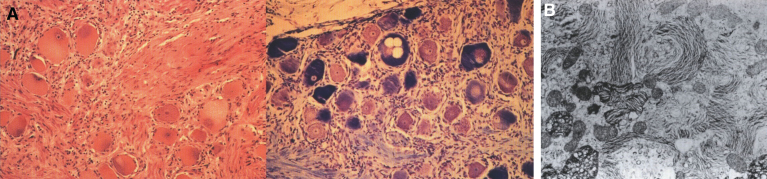
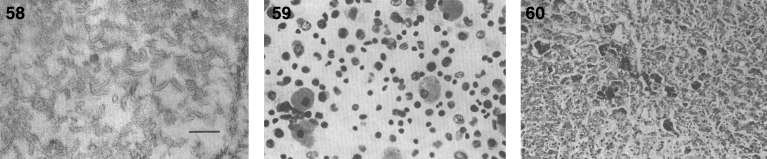
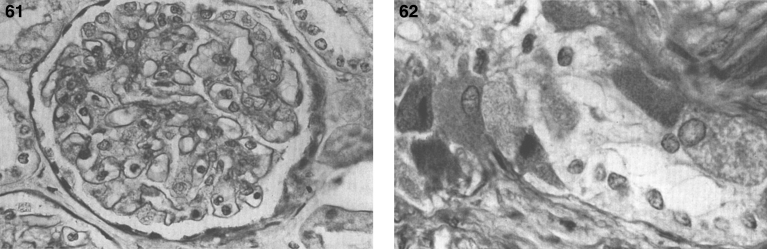
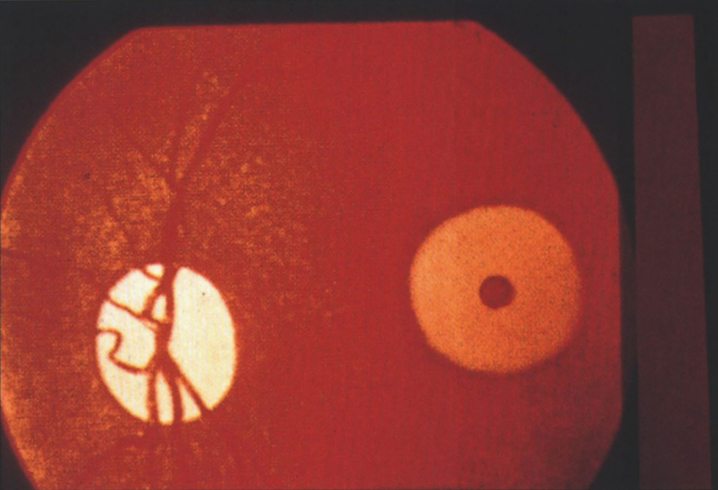
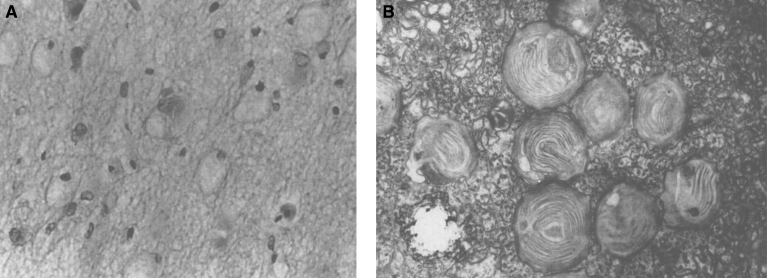
Lysosomes are cytoplasmic organelles that contain a variety of different hydrolases. A genetic deficiency in the enzymatic activity of one of these hydrolases will lead to the accumulation of the material meant for lysosomal degradation. Examples include glycogen in the case of Pompe disease, glycosaminoglycans in the case of the mucopolysaccharidoses, glycoproteins in the cases of the oligosaccharidoses, and sphingolipids in the cases of Niemann-Pick disease types A and B, Gaucher disease, Tay-Sachs disease, Krabbe disease, and metachromatic leukodystrophy. Sometimes, the lysosomal storage can be caused not by the enzymatic deficiency of one of the hydrolases, but by the deficiency of an activator protein, as occurs in the AB variant of GM2 gangliosidosis. Still other times, the accumulated lysosomal material results from failed egress of a small molecule as a consequence of a deficient transporter, as in cystinosis or Salla disease. In the last couple of decades, enzyme replacement therapy has become available for a number of lysosomal storage diseases. Examples include imiglucerase, taliglucerase and velaglucerase for Gaucher disease, laronidase for Hurler disease, idursulfase for Hunter disease, elosulfase for Morquio disease, galsulfase for Maroteaux-Lamy disease, alglucosidase alfa for Pompe disease, and agalsidase alfa and beta for Fabry disease. In addition, substrate reduction therapy has been approved for certain disorders, such as eliglustat for Gaucher disease. The advent of treatment options for some of these disorders has led to newborn screening pilot studies, and ultimately to the addition of Pompe disease and Hurler disease to the Recommended Uniform Screening Panel (RUSP) in 2015 and 2016, respectively.
Keywords: Fabry disease; Farber disease; GM1 gangliosidosis; Gaucher disease; Krabbe disease; Lysosomal storage diseases; Niemann-Pick disease; Sandhoff disease; Schindler disease; Tay-Sachs disease; cystinosis; free sialic acid storage disease; metachromatic leukodystrophy; mucolipidosis IV; newborn screening.
Figures

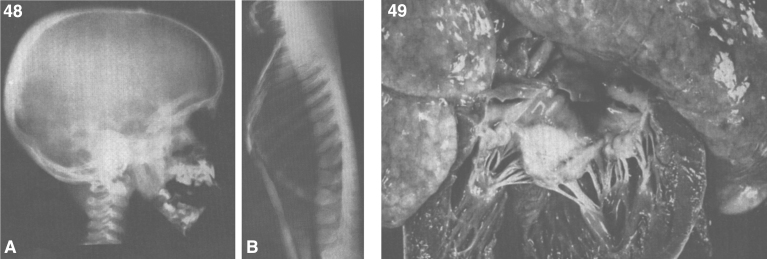
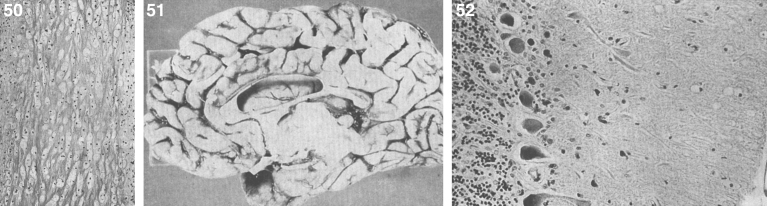
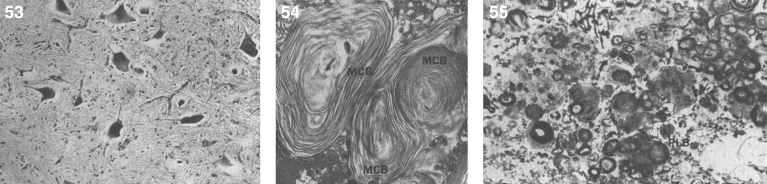
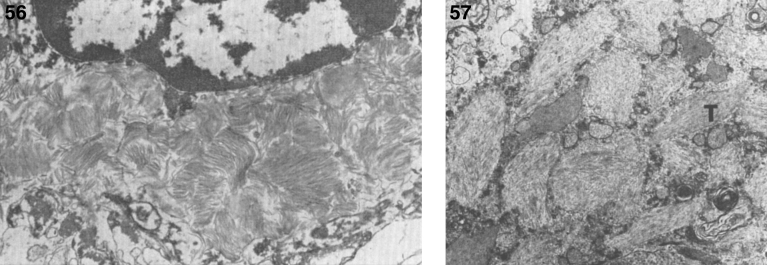
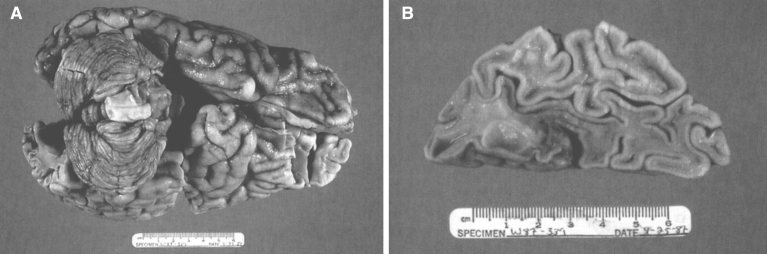
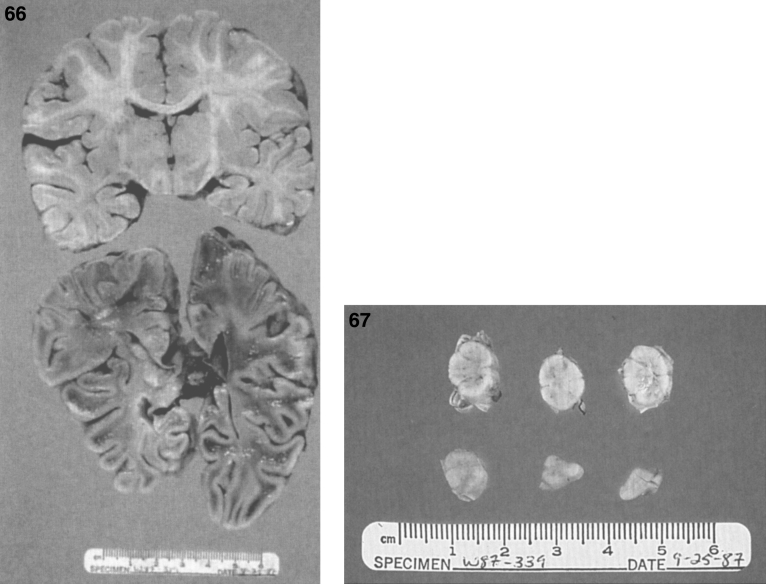
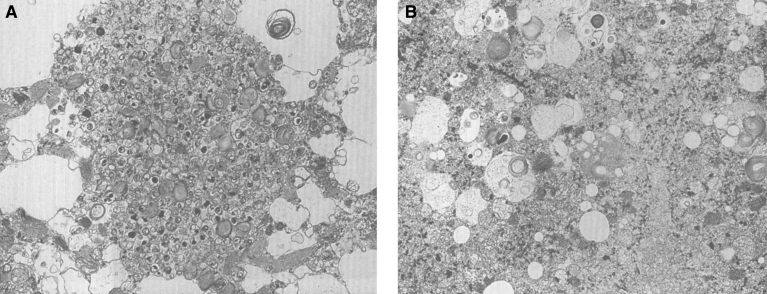
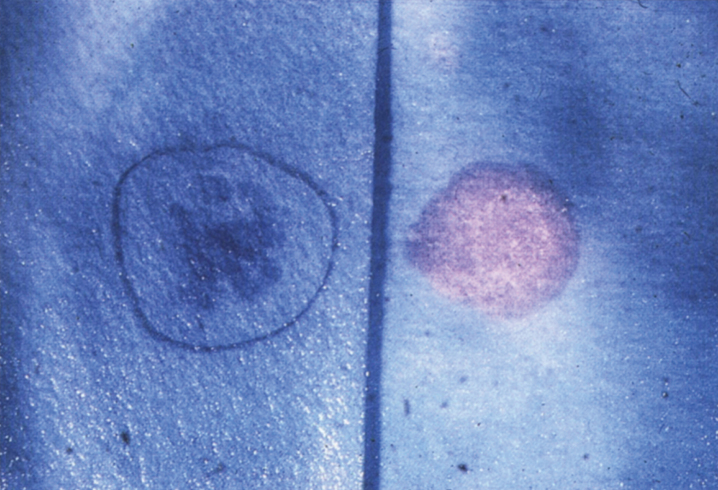
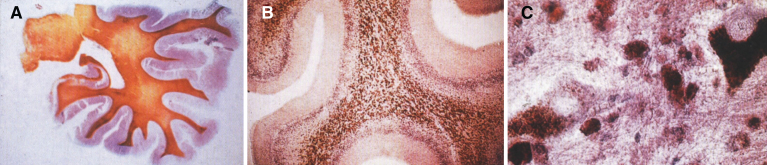
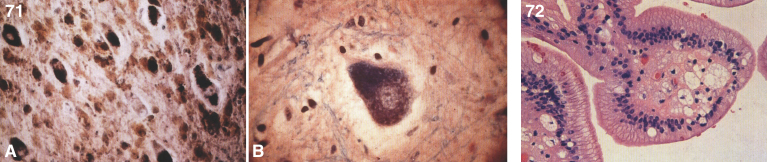
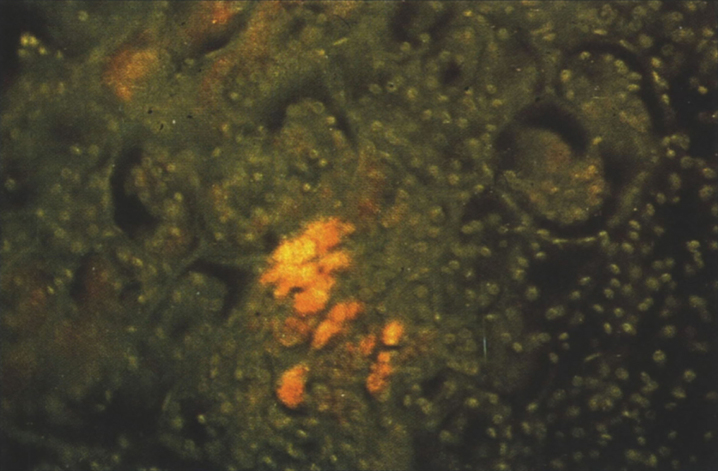
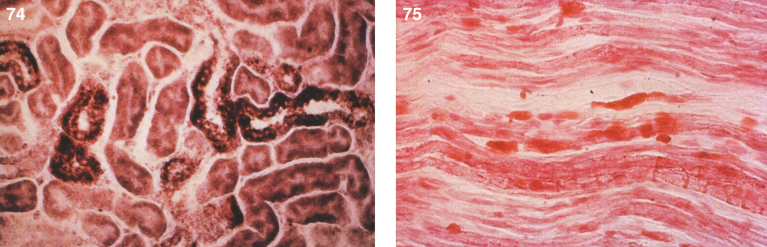
References
-
- Sabatini D.D. and Adesnik M.B., The Biogenesis of Membranes and Organelles In: Valle DL, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; 2014. [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102899807
-
- Bowers W.E., Christian de Duve and the discovery of lysosomes and peroxisomes, Trends Cell Biol 8(8) (1998), 330–333. - PubMed
-
- Passarge E., Color Atlas of Genetics Thieme Publishing Group; 1995.
-
- Coutinho M.F., Prata M.J. and Alves S., A shortcut to the lysosome: The mannose-6-phosphate-independent pathway, Mol Genet Metab 107(3) (2012), 257–266. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources