

Efficient Generation of Transcriptomic Profiles by Random Composite Measurements
- PMID: 29153835
- PMCID: PMC5726792
- DOI: 10.1016/j.cell.2017.10.023
Efficient Generation of Transcriptomic Profiles by Random Composite Measurements
Abstract
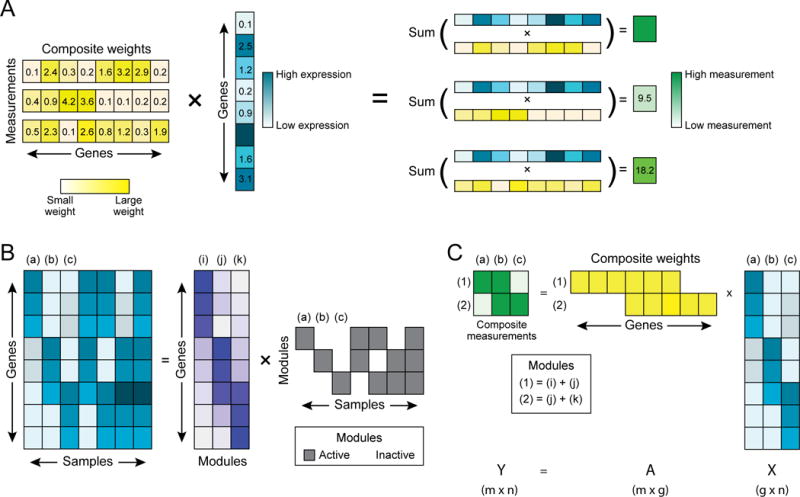
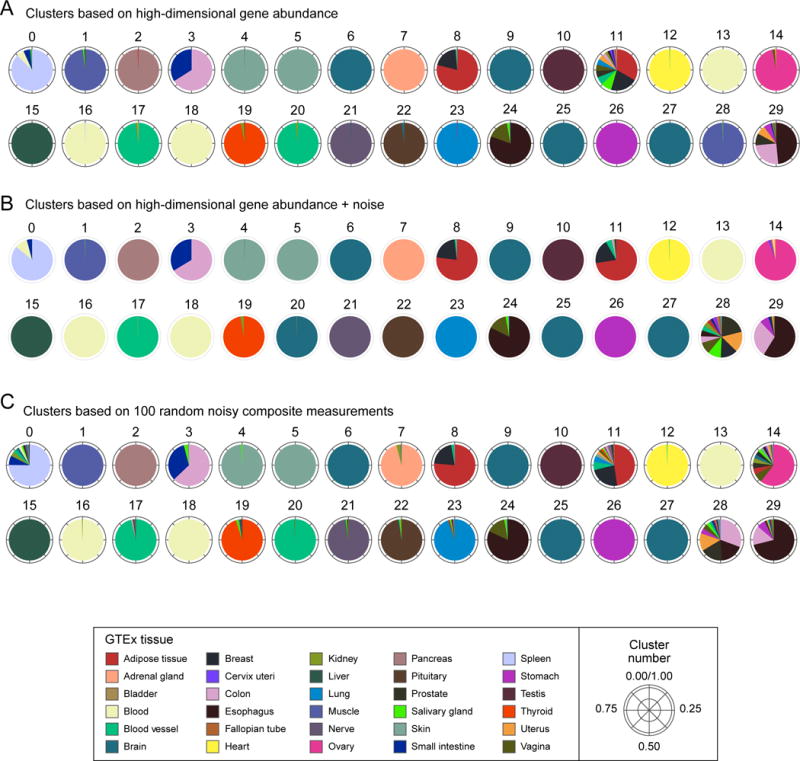
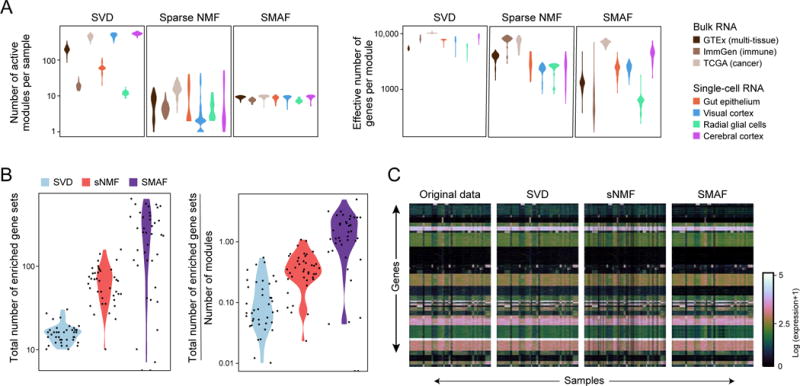
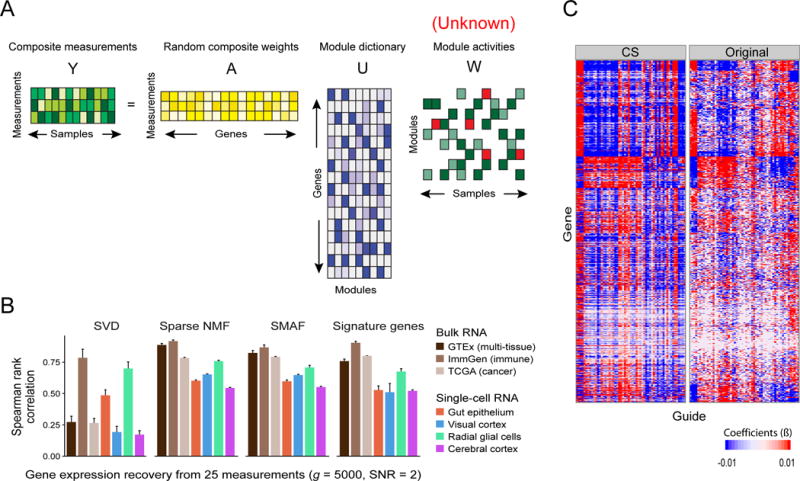
RNA profiles are an informative phenotype of cellular and tissue states but can be costly to generate at massive scale. Here, we describe how gene expression levels can be efficiently acquired with random composite measurements-in which abundances are combined in a random weighted sum. We show (1) that the similarity between pairs of expression profiles can be approximated with very few composite measurements; (2) that by leveraging sparse, modular representations of gene expression, we can use random composite measurements to recover high-dimensional gene expression levels (with 100 times fewer measurements than genes); and (3) that it is possible to blindly recover gene expression from composite measurements, even without access to training data. Our results suggest new compressive modalities as a foundation for massive scaling in high-throughput measurements and new insights into the interpretation of high-dimensional data.
Keywords: compressed sensing; gene expression; random composite measurements.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Aghagolzadeh M, Radha H. New Guarantees for Blind Compressed Sensing. 2015:1227–1234.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
