

Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications
- PMID: 29154141
- PMCID: PMC5901717
- DOI: 10.1016/S1474-4422(17)30401-5
Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications
Abstract
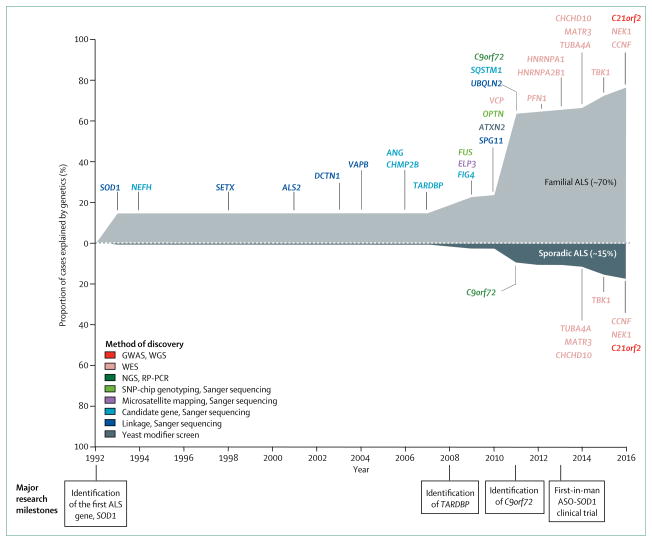
Background: The disease course of amyotrophic lateral sclerosis (ALS) is rapid and, because its pathophysiology is unclear, few effective treatments are available. Genetic research aims to understand the underlying mechanisms of ALS and identify potential therapeutic targets. The first gene associated with ALS was SOD1, identified in 1993 and, by early 2014, more than 20 genes had been identified as causative of, or highly associated with, ALS. These genetic discoveries have identified key disease pathways that are therapeutically testable and could potentially lead to the development of better treatments for people with ALS.
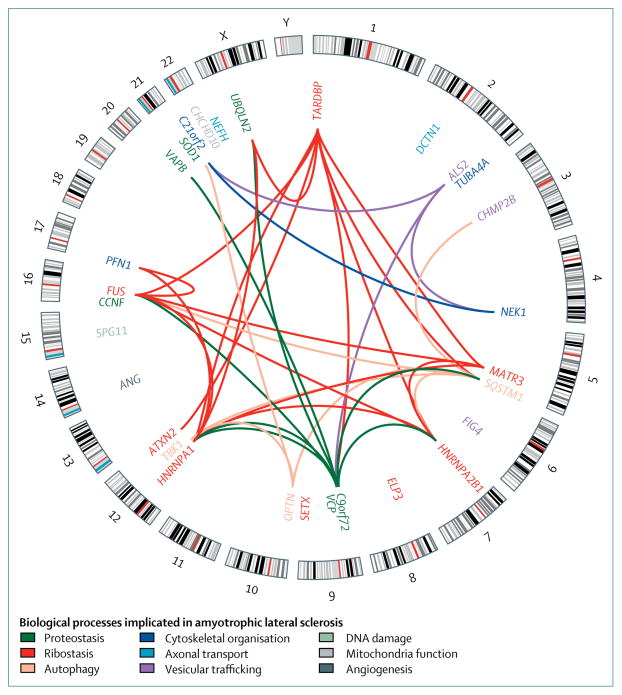
Recent developments: Since 2014, seven additional genes have been associated with ALS (MATR3, CHCHD10, TBK1, TUBA4A, NEK1, C21orf2, and CCNF), all of which were identified by genome-wide association studies, whole genome studies, or exome sequencing technologies. Each of the seven novel genes code for proteins associated with one or more molecular pathways known to be involved in ALS. These pathways include dysfunction in global protein homoeostasis resulting from abnormal protein aggregation or a defect in the protein clearance pathway, mitochondrial dysfunction, altered RNA metabolism, impaired cytoskeletal integrity, altered axonal transport dynamics, and DNA damage accumulation due to defective DNA repair. Because these novel genes share common disease pathways with other genes implicated in ALS, therapeutics targeting these pathways could be useful for a broad group of patients stratified by genotype. However, the effects of these novel genes have not yet been investigated in animal models, which will be a key step to translating these findings into clinical practice. WHERE NEXT?: The identification of these seven novel genes has been important in unravelling the molecular mechanisms underlying ALS. However, our understanding of what causes ALS is not complete, and further genetic research will provide additional detail about its causes. Increased genetic knowledge will also identify potential therapeutic targets and could lead to the development of individualised medicine for patients with ALS. These developments will have a direct effect on clinical practice when genome sequencing becomes a routine and integral part of disease diagnosis and management.
Copyright © 2018 Elsevier Ltd. All rights reserved.
Conflict of interest statement
AC received personal fees from Biogen Idec, Mitsubishi, and Neuraltus. BJT received grants from Merck, Microsoft Research, the Amyotrophic Lateral Sclerosis Association, the Packard Center for Amyotrophic Lateral Sclerosis Research, the Muscular Dystrophy Association, the Center for Disease Prevention and Control, the Myasthenia Gravis Foundation, the Italian Football Association, and the Italian Amyotrophic Lateral Sclerosis Association; and holds the European patent (16204118.0-1404) on the clinical testing and therapeutic intervention for the hexanucleotide repeat expansion of
Figures
References
-
- Talbott EO, Malek AM, Lacomis D. The epidemiology of amyotrophic lateral sclerosis. Handb Clin Neurol. 2016;138:225–38. - PubMed
-
- Blasco H, Patin F, Andres CR, Corcia P, Gordon PH. Amyotrophic lateral sclerosis, 2016: existing therapies and the ongoing search for neuroprotection. Expert Opin Pharmacother. 2016;17:1669–82. - PubMed
-
- Writing Group, Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16:505–12. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
