

Mitochondria-targeted ubiquinone (MitoQ) enhances acetaldehyde clearance by reversing alcohol-induced posttranslational modification of aldehyde dehydrogenase 2: A molecular mechanism of protection against alcoholic liver disease
- PMID: 29156373
- PMCID: PMC5700831
- DOI: 10.1016/j.redox.2017.11.005
Mitochondria-targeted ubiquinone (MitoQ) enhances acetaldehyde clearance by reversing alcohol-induced posttranslational modification of aldehyde dehydrogenase 2: A molecular mechanism of protection against alcoholic liver disease
Abstract
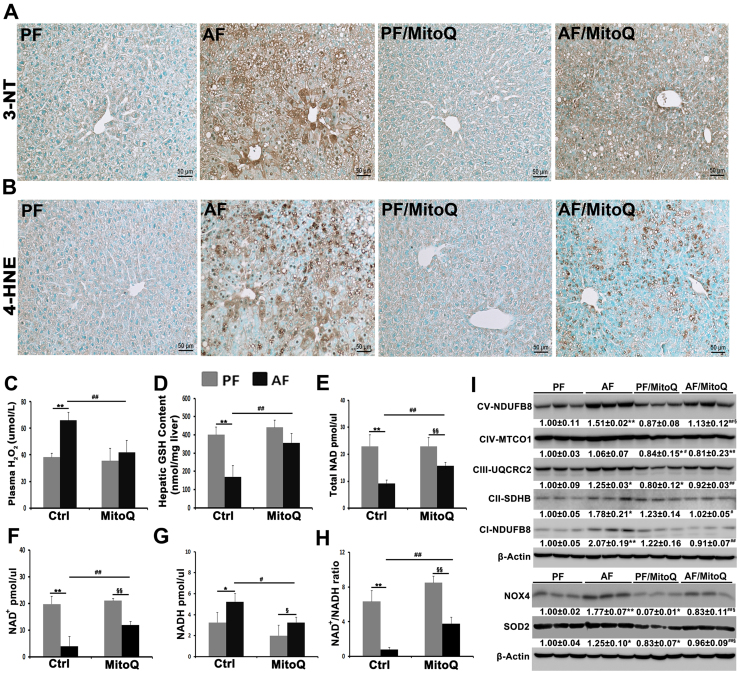
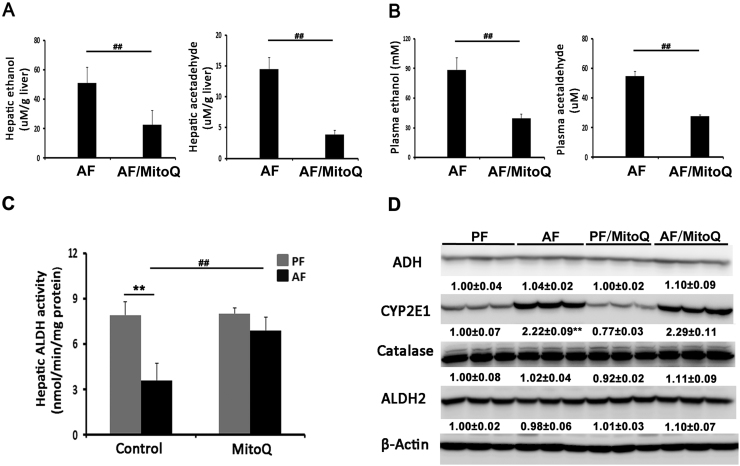
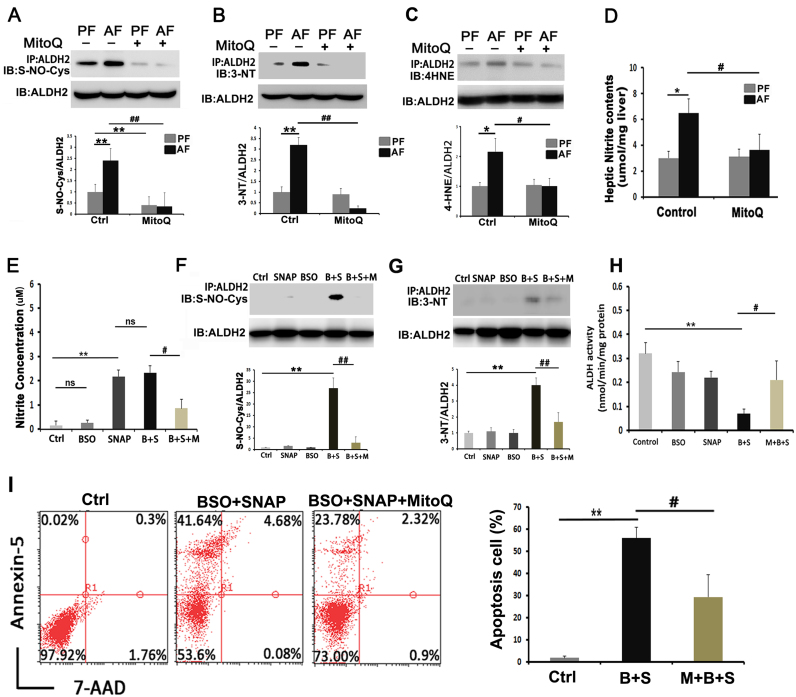
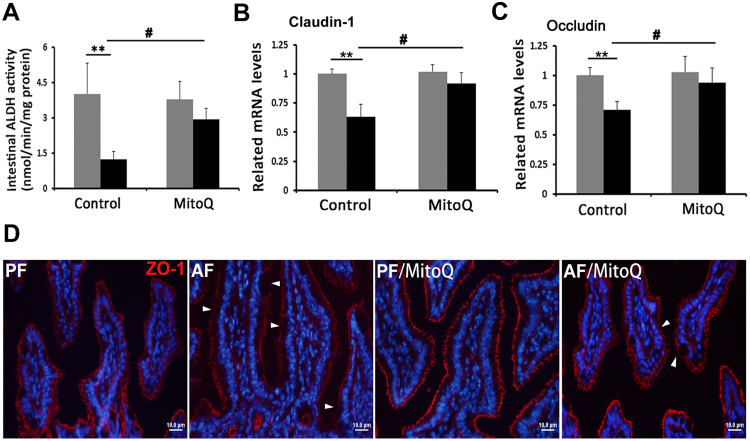
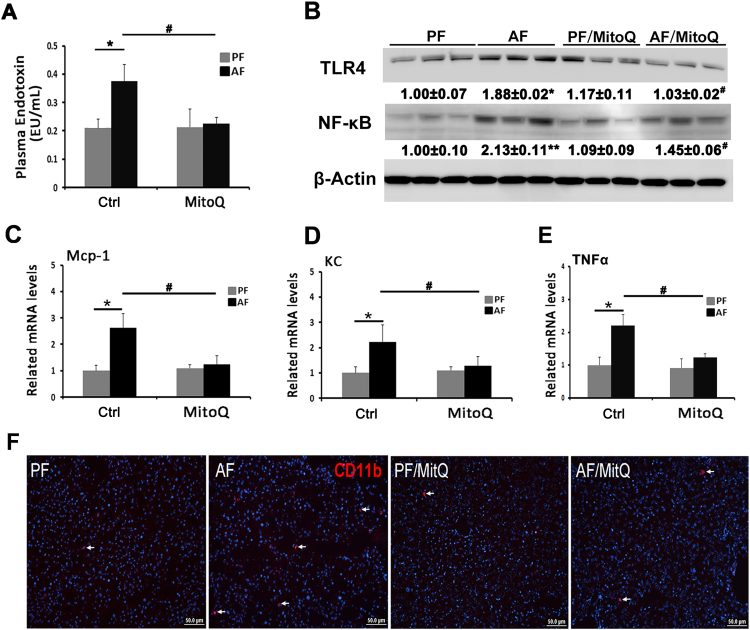
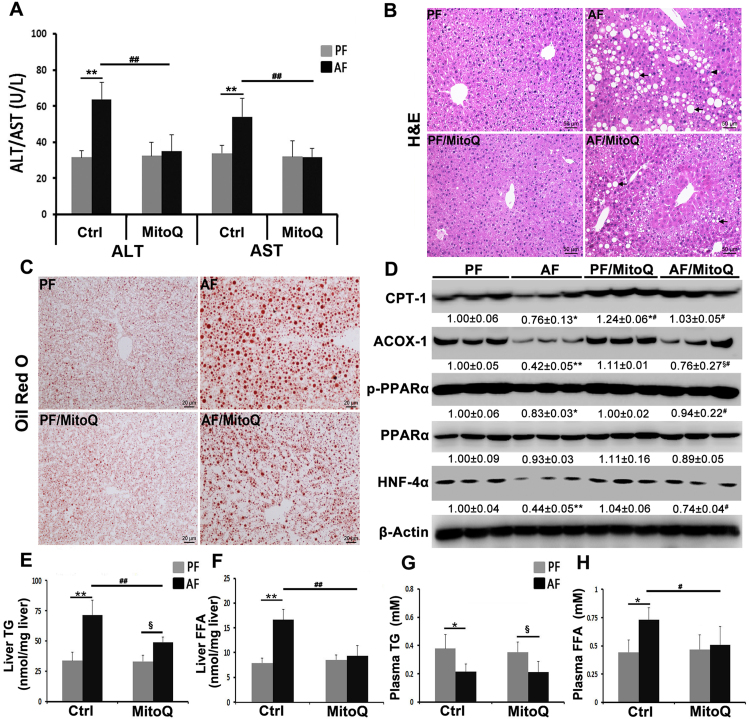
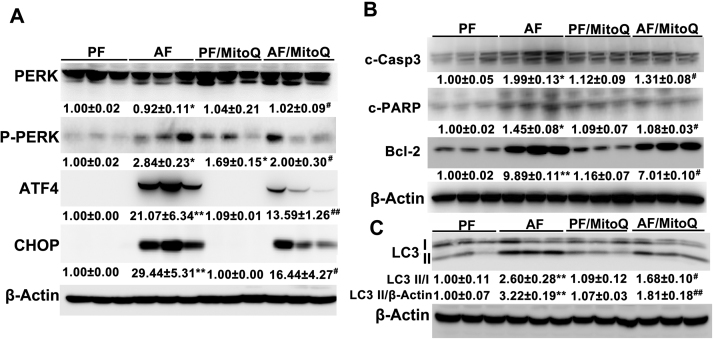
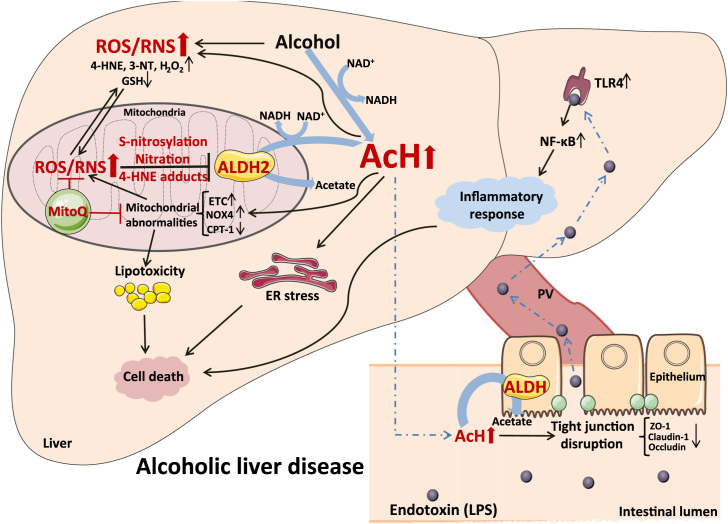
Alcohol metabolism in the liver generates highly toxic acetaldehyde. Breakdown of acetaldehyde by aldehyde dehydrogenase 2 (ALDH2) in the mitochondria consumes NAD+ and generates reactive oxygen/nitrogen species, which represents a fundamental mechanism in the pathogenesis of alcoholic liver disease (ALD). A mitochondria-targeted lipophilic ubiquinone (MitoQ) has been shown to confer greater protection against oxidative damage in the mitochondria compared to untargeted antioxidants. The present study aimed to investigate if MitoQ could preserve mitochondrial ALDH2 activity and speed up acetaldehyde clearance, thereby protects against ALD. Male C57BL/6J mice were exposed to alcohol for 8 weeks with MitoQ supplementation (5mg/kg/d) for the last 4 weeks. MitoQ ameliorated alcohol-induced oxidative/nitrosative stress and glutathione deficiency. It also reversed alcohol-reduced hepatic ALDH activity and accelerated acetaldehyde clearance through modulating ALDH2 cysteine S-nitrosylation, tyrosine nitration and 4-hydroxynonenol adducts formation. MitoQ ameliorated nitric oxide (NO) donor-mediated ADLH2 S-nitrosylation and nitration in Hepa-1c1c7 cells under glutathion depletion condition. In addition, alcohol-increased circulating acetaldehyde levels were accompanied by reduced intestinal ALDH activity and impaired intestinal barrier. In accordance, MitoQ reversed alcohol-increased plasma endotoxin levels and hepatic toll-like receptor 4 (TLR4)-NF-κB signaling along with subsequent inhibition of inflammatory cell infiltration. MitoQ also reversed alcohol-induced hepatic lipid accumulation through enhancing fatty acid β-oxidation. Alcohol-induced ER stress and apoptotic cell death signaling were reversed by MitoQ. This study demonstrated that speeding up acetaldehyde clearance by preserving ALDH2 activity critically mediates the beneficial effect of MitoQ on alcohol-induced pathogenesis at the gut-liver axis.
Keywords: Alcoholic liver disease; Aldehyde dehydrogenase 2; MitoQ; Posttranslational modification.
Copyright © 2017 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Masarone M., Rosato V., Dallio M., Abenavoli L., Federico A., Loguercio C., Persico M. Epidemiology and Natural History of Alcoholic Liver Disease. Rev. Recent Clin. Trials. 2016;11(3):167–174. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
