

Nitro-fatty acid inhibition of triple-negative breast cancer cell viability, migration, invasion, and tumor growth
- PMID: 29158255
- PMCID: PMC5787792
- DOI: 10.1074/jbc.M117.814368
Nitro-fatty acid inhibition of triple-negative breast cancer cell viability, migration, invasion, and tumor growth
Abstract
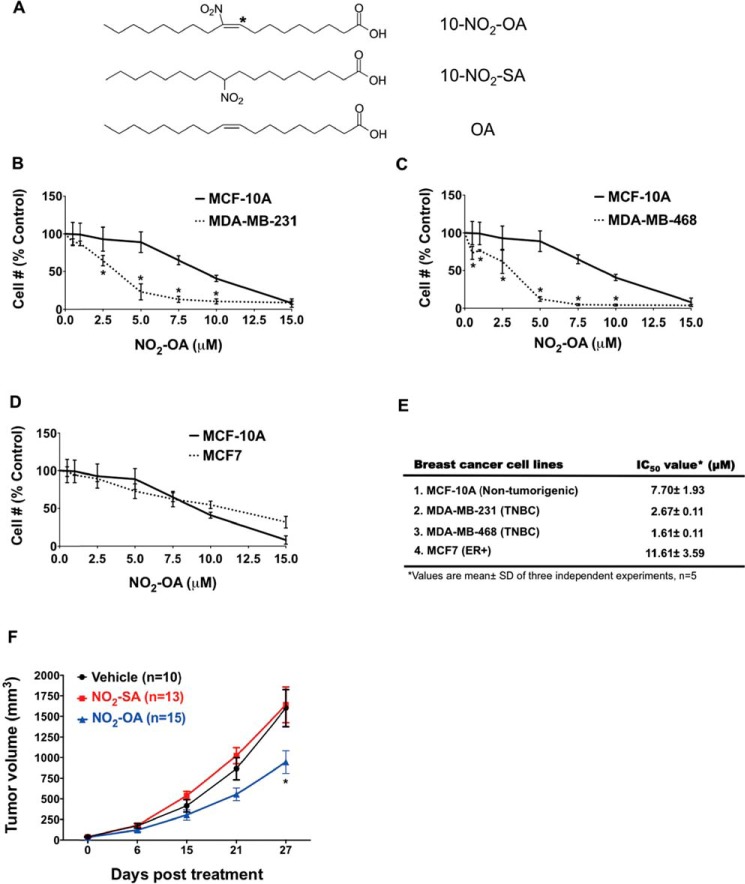
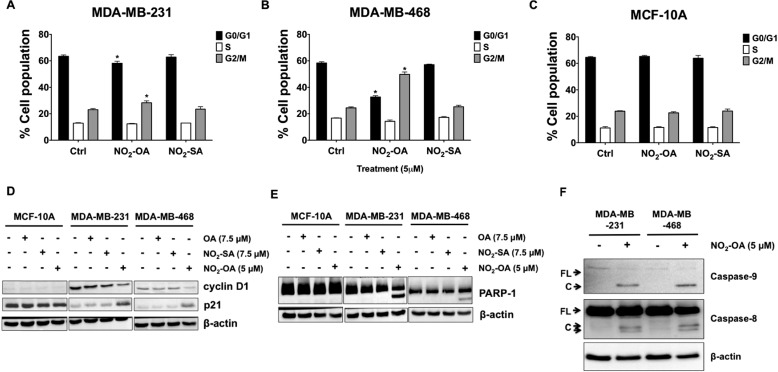
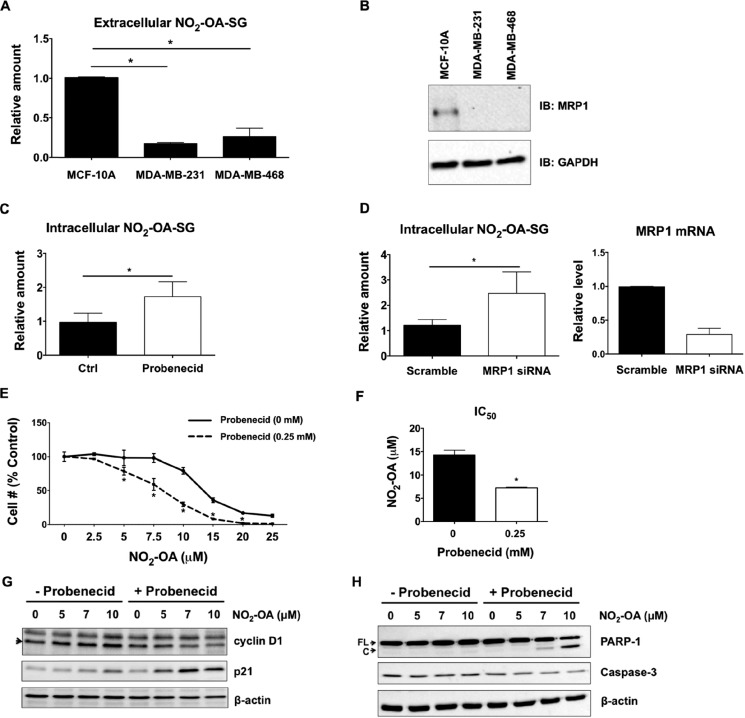
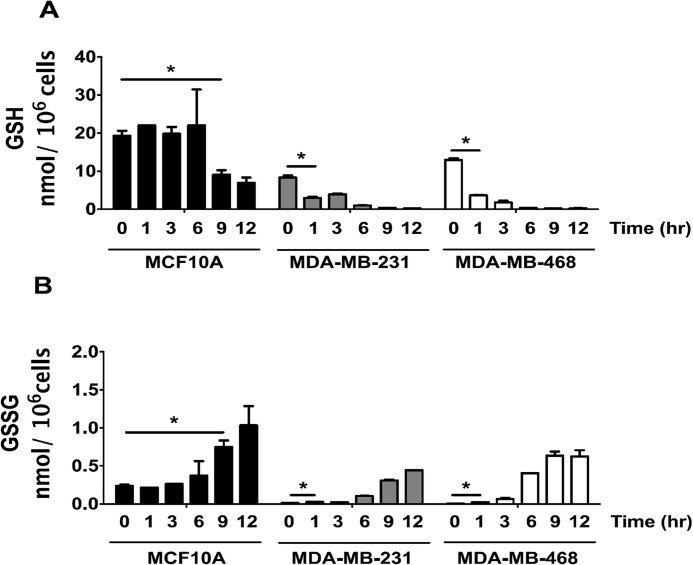
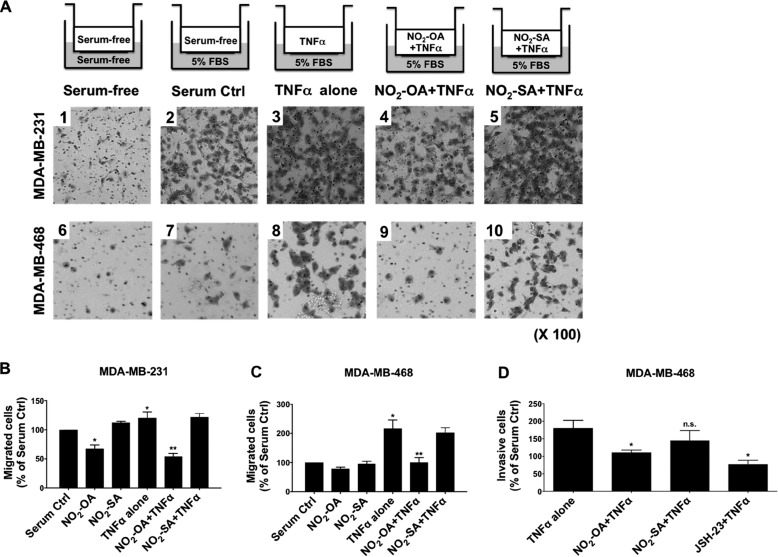
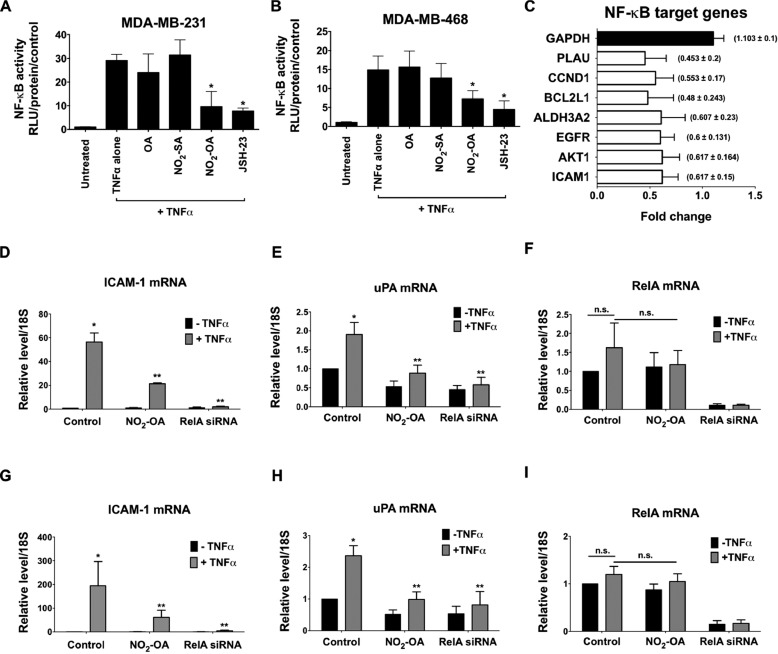
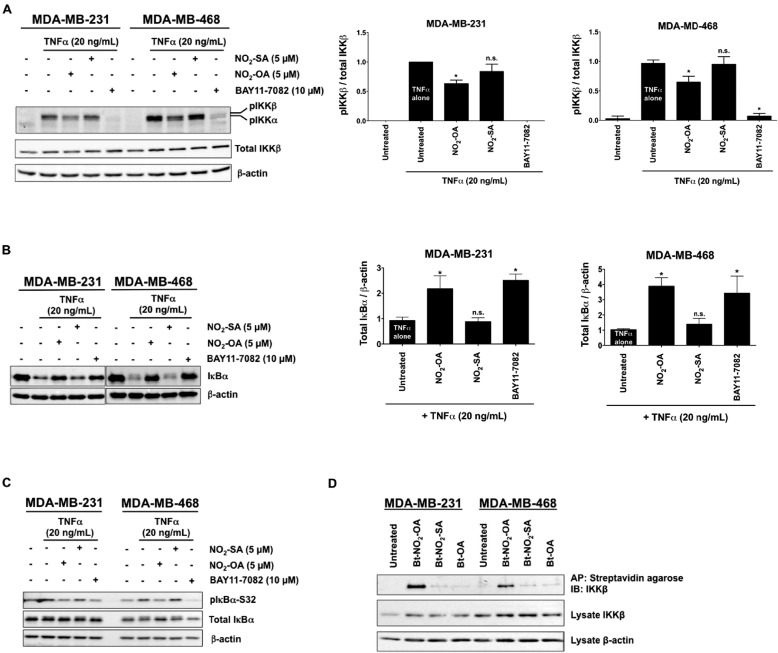
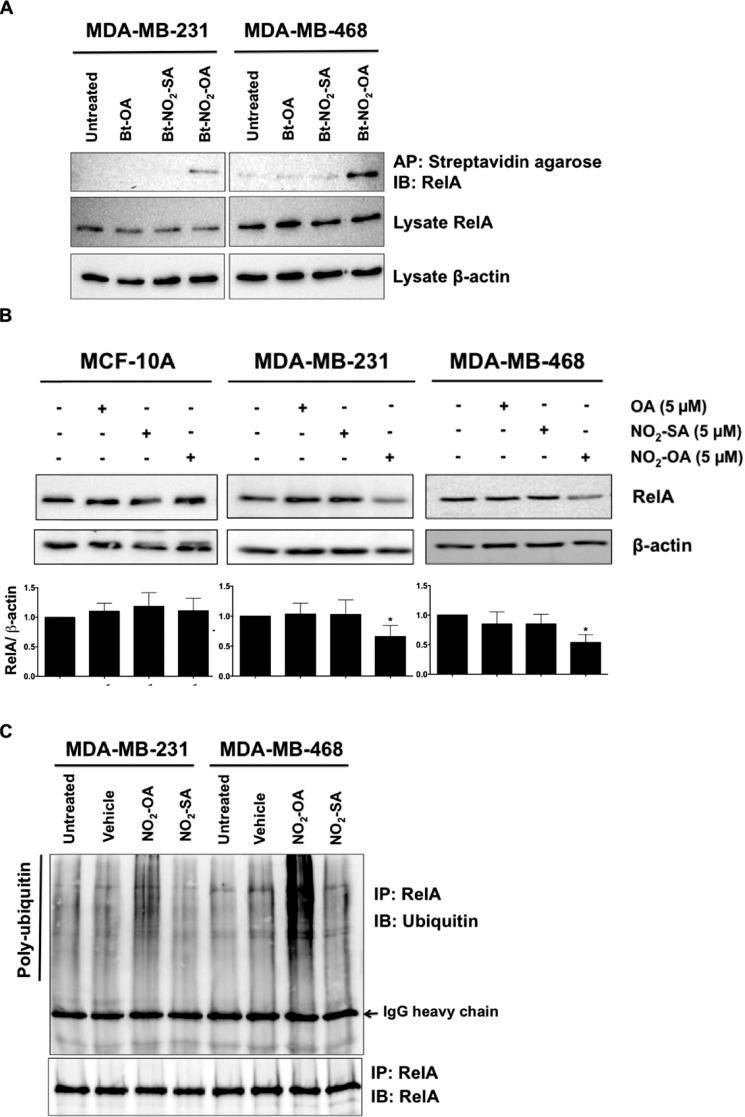
Triple-negative breast cancer (TNBC) comprises ∼20% of all breast cancers and is the most aggressive mammary cancer subtype. Devoid of the estrogen and progesterone receptors, along with the receptor tyrosine kinase ERB2 (HER2), that define most mammary cancers, there are no targeted therapies for patients with TNBC. This, combined with a high metastatic rate and a lower 5-year survival rate than for other breast cancer phenotypes, means there is significant unmet need for new therapeutic strategies. Herein, the anti-neoplastic effects of the electrophilic fatty acid nitroalkene derivative, 10-nitro-octadec-9-enoic acid (nitro-oleic acid, NO2-OA), were investigated in multiple preclinical models of TNBC. NO2-OA reduced TNBC cell growth and viability in vitro, attenuated TNFα-induced TNBC cell migration and invasion, and inhibited the tumor growth of MDA-MB-231 TNBC cell xenografts in the mammary fat pads of female nude mice. The up-regulation of these aggressive tumor cell growth, migration, and invasion phenotypes is mediated in part by the constitutive activation of pro-inflammatory nuclear factor κB (NF-κB) signaling in TNBC. NO2-OA inhibited TNFα-induced NF-κB transcriptional activity in human TNBC cells and suppressed downstream NF-κB target gene expression, including the metastasis-related proteins intercellular adhesion molecule-1 and urokinase-type plasminogen activator. The mechanisms accounting for NF-κB signaling inhibition by NO2-OA in TNBC cells were multifaceted, as NO2-OA (a) inhibited the inhibitor of NF-κB subunit kinase β phosphorylation and downstream inhibitor of NF-κB degradation, (b) alkylated the NF-κB RelA protein to prevent DNA binding, and (c) promoted RelA polyubiquitination and proteasomal degradation. Comparisons with non-tumorigenic human breast epithelial MCF-10A and MCF7 cells revealed that NO2-OA more selectively inhibited TNBC function. This was attributed to more facile mechanisms for maintaining redox homeostasis in normal breast epithelium, including a more favorable thiol/disulfide balance, greater extents of multidrug resistance protein-1 (MRP1) expression, and greater MRP1-mediated efflux of NO2-OA-glutathione conjugates. These observations reveal that electrophilic fatty acid nitroalkenes react with more alkylation-sensitive targets in TNBC cells to inhibit growth and viability.
Keywords: NF-kappaB; breast cancer; cancer chemoprevention; drug action; proliferation; reactive nitrogen species (RNS); reactive oxygen species (ROS); signaling; tumor immunology; tumor necrosis factor (TNF).
© 2018 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
B. A. F., S. G. W., S. R. W., and C. C. W. acknowledge interest in Complexa, Inc. No potential conflicts of interest were disclosed by other authors
Figures
References
-
- Alexander R. L., Bates D. J., Wright M. W., King S. B., and Morrow C. S. (2006) Modulation of nitrated lipid signaling by multidrug resistance protein 1 (MRP1): glutathione conjugation and MRP1-mediated efflux inhibit nitrolinoleic acid-induced, PPARγ-dependent transcription activation. Biochemistry 45, 7889–7896 10.1021/bi0605639 - DOI - PubMed
-
- Bauer K. R., Brown M., Cress R. D., Parise C. A., and Caggiano V. (2007) Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype. Cancer 109, 1721–1728 10.1002/cncr.22618 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
