

Evaluation of the template-based modeling in CASP12
- PMID: 29159950
- PMCID: PMC5877821
- DOI: 10.1002/prot.25425
Evaluation of the template-based modeling in CASP12
Abstract
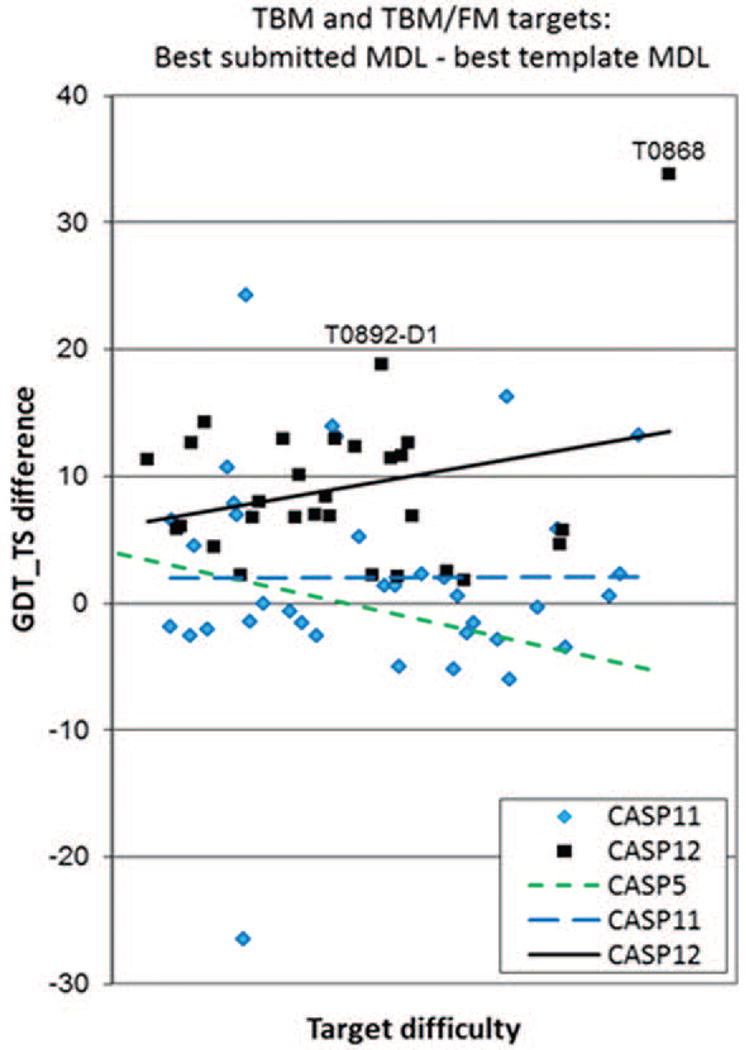
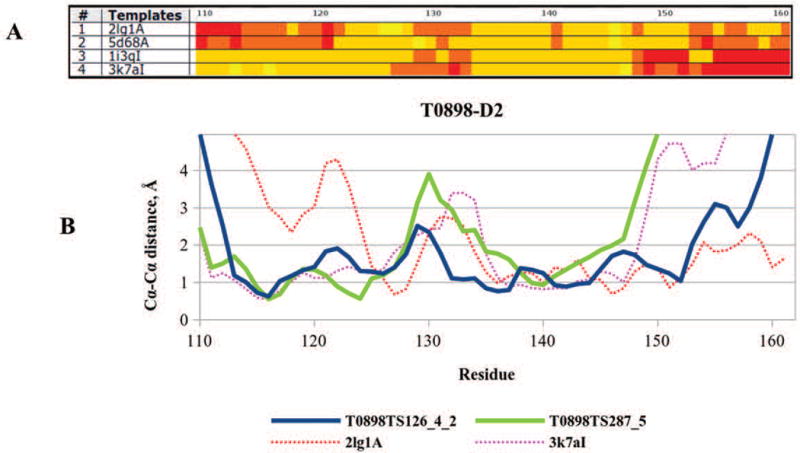
The article describes results of numerical evaluation of CASP12 models submitted on targets for which structural templates could be identified and for which servers produced models of relatively high accuracy. The emphasis is on analysis of details of models, and how well the models compete with experimental structures. Performance of contributing research groups is measured in terms of backbone accuracy, all-atom local geometry, and the ability to estimate local errors in models. Separate analyses for all participating groups and automatic servers were carried out. Compared with the last CASP, two years ago, there have been significant improvements in a number of areas, particularly the accuracy of protein backbone atoms, accuracy of sequence alignment between models and available structures, increased accuracy over that which can be obtained from simple copying of a closest template, and accuracy of modeling of sub-structures not present in the closest template. These advancements are likely associated with more effective strategies to build non-template regions of the targets ab initio, better algorithms to combine information from multiple templates, enhanced refinement methods, and better methods for estimating model accuracy.
Keywords: CASP; high accuracy models; numerical evaluation measures; protein structure prediction; template-based protein modeling.
© 2017 Wiley Periodicals, Inc.
Figures







References
-
- Zemla A, Venclovas, Moult J, Fidelis K. Processing and evaluation of predictions in CASP4. Proteins. 2001;(Suppl 5):13–21. - PubMed
