

ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease
- PMID: 29162642
- PMCID: PMC5749174
- DOI: 10.1101/gr.226621.117
ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease
Abstract
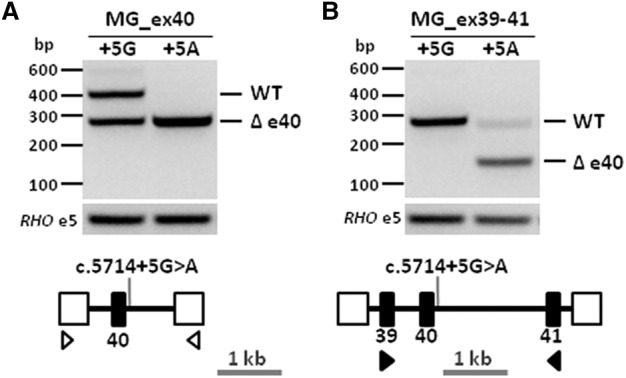
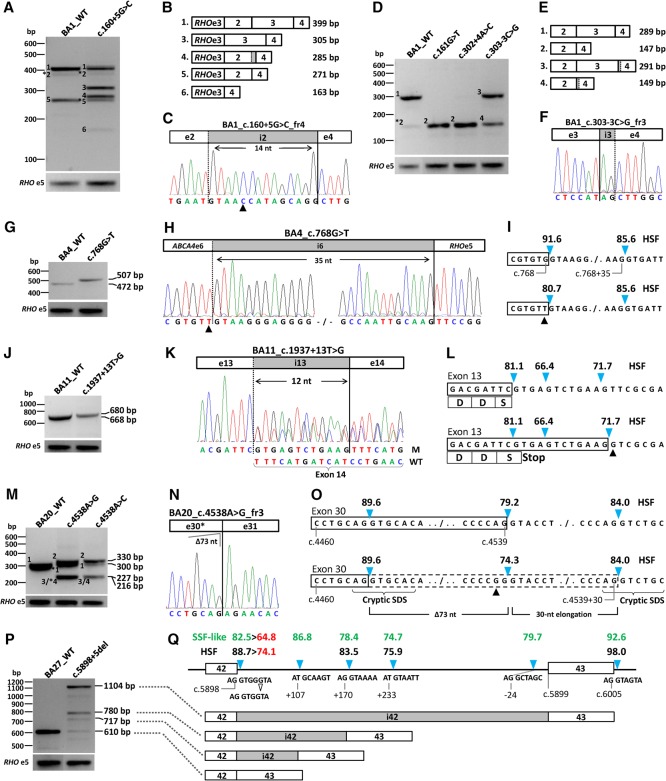
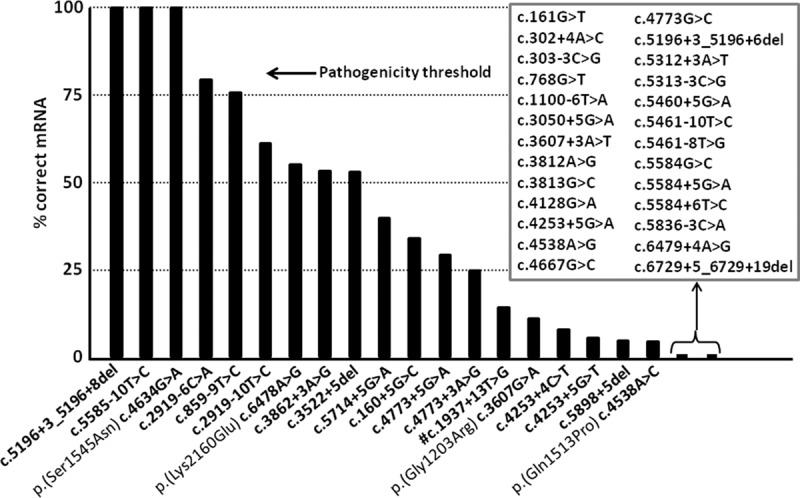
Stargardt disease is caused by variants in the ABCA4 gene, a significant part of which are noncanonical splice site (NCSS) variants. In case a gene of interest is not expressed in available somatic cells, small genomic fragments carrying potential disease-associated variants are tested for splice abnormalities using in vitro splice assays. We recently discovered that when using small minigenes lacking the proper genomic context, in vitro results do not correlate with splice defects observed in patient cells. We therefore devised a novel strategy in which a bacterial artificial chromosome was employed to generate midigenes, splice vectors of varying lengths (up to 11.7 kb) covering almost the entire ABCA4 gene. These midigenes were used to analyze the effect of all 44 reported and three novel NCSS variants on ABCA4 pre-mRNA splicing. Intriguingly, multi-exon skipping events were observed, as well as exon elongation and intron retention. The analysis of all reported NCSS variants in ABCA4 allowed us to reveal the nature of aberrant splicing events and to classify the severity of these mutations based on the residual fraction of wild-type mRNA. Our strategy to generate large overlapping splice vectors carrying multiple exons, creating a toolbox for robust and high-throughput analysis of splice variants, can be applied to all human genes.
© 2018 Sangermano et al.; Published by Cold Spring Harbor Laboratory Press.
Figures

References
-
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, et al. 1997. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 15: 236–246. - PubMed
-
- Aukrust I, Jansson RW, Bredrup C, Rusaas HE, Berland S, Jorgensen A, Haug MG, Rodahl E, Houge G, Knappskog PM. 2017. The intronic ABCA4 c.5461–10T>C variant, frequently seen in patients with Stargardt disease, causes splice defects and reduced ABCA4 protein level. Acta Ophthalmol 95: 240–246. - PubMed
-
- Baralle M, Skoko N, Knezevich A, De Conti L, Motti D, Bhuvanagiri M, Baralle D, Buratti E, Baralle FE. 2006. NF1 mRNA biogenesis: effect of the genomic milieu in splicing regulation of the NF1 exon 37 region. FEBS Lett 580: 4449–4456. - PubMed
-
- Blacharski PA. 1988. Fundus flavimaculatus. In Retinal dystrophies and degenerations (ed. Newsome DA), pp. 135–159. Raven Press, New York.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical