

Structural basis for the antiarrhythmic blockade of a potassium channel with a small molecule
- PMID: 29162702
- PMCID: PMC5893172
- DOI: 10.1096/fj.201700349R
Structural basis for the antiarrhythmic blockade of a potassium channel with a small molecule
Abstract
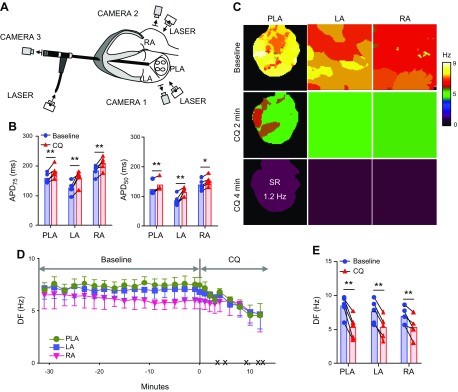
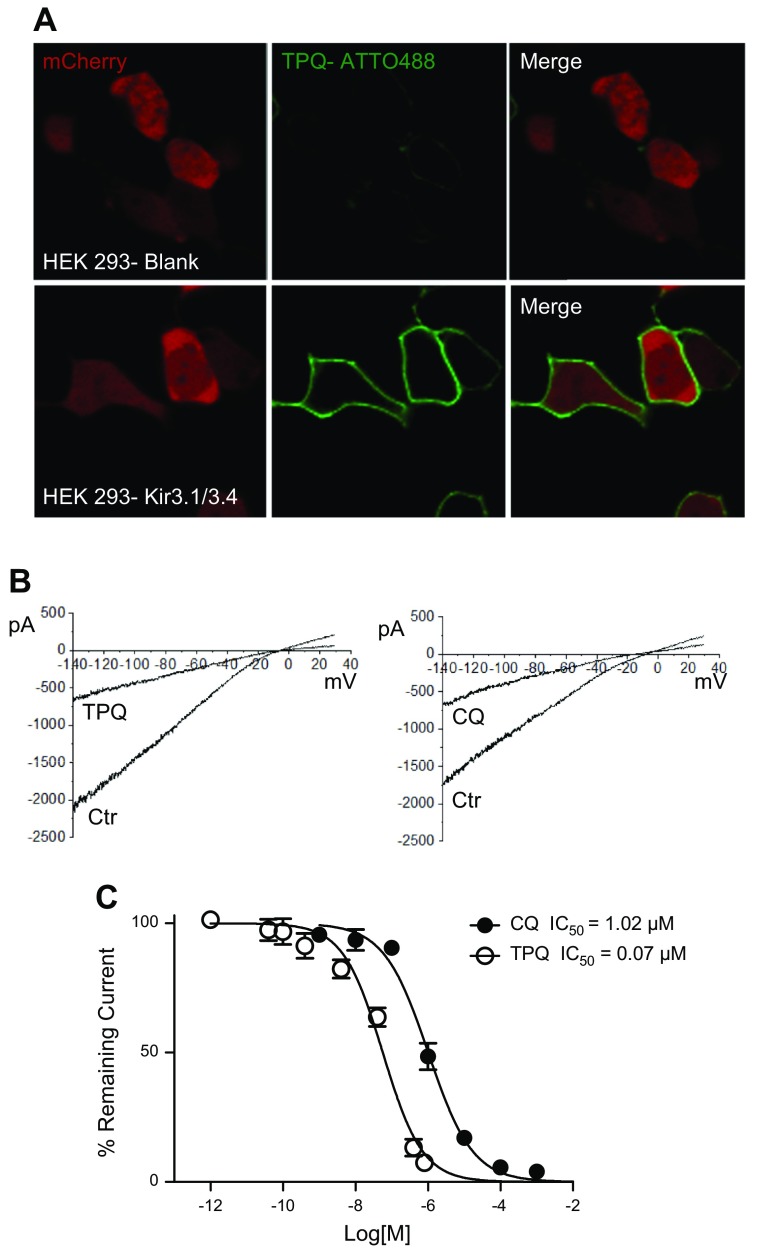
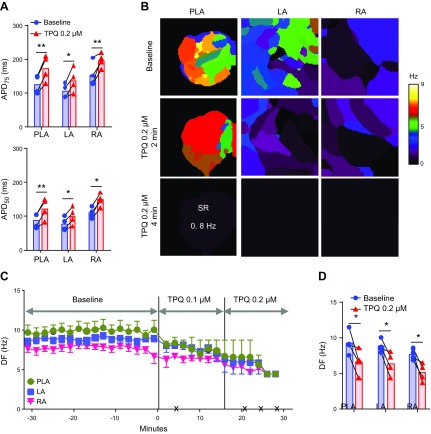
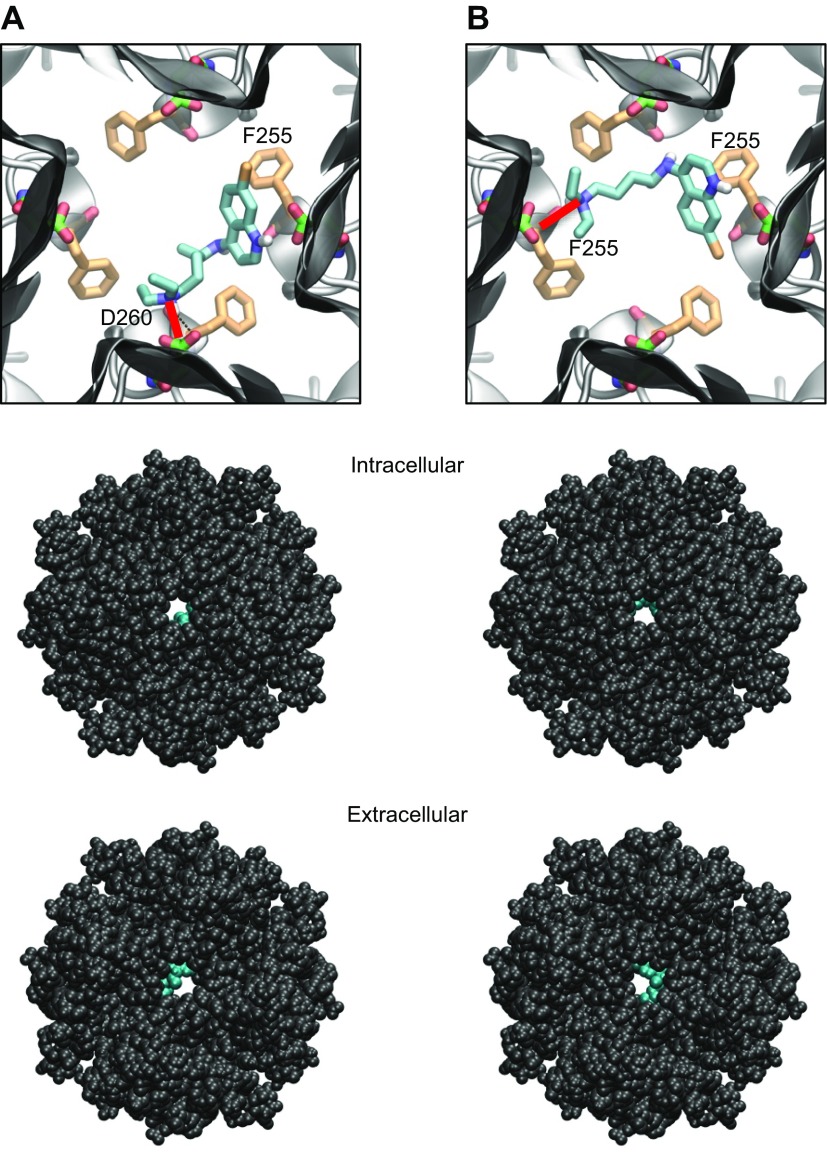
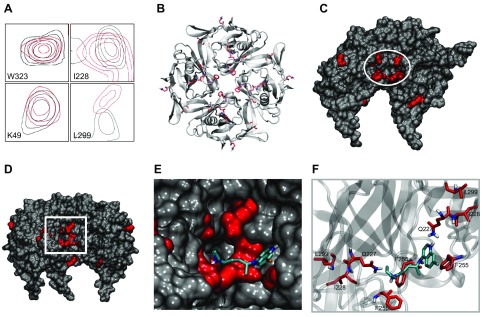
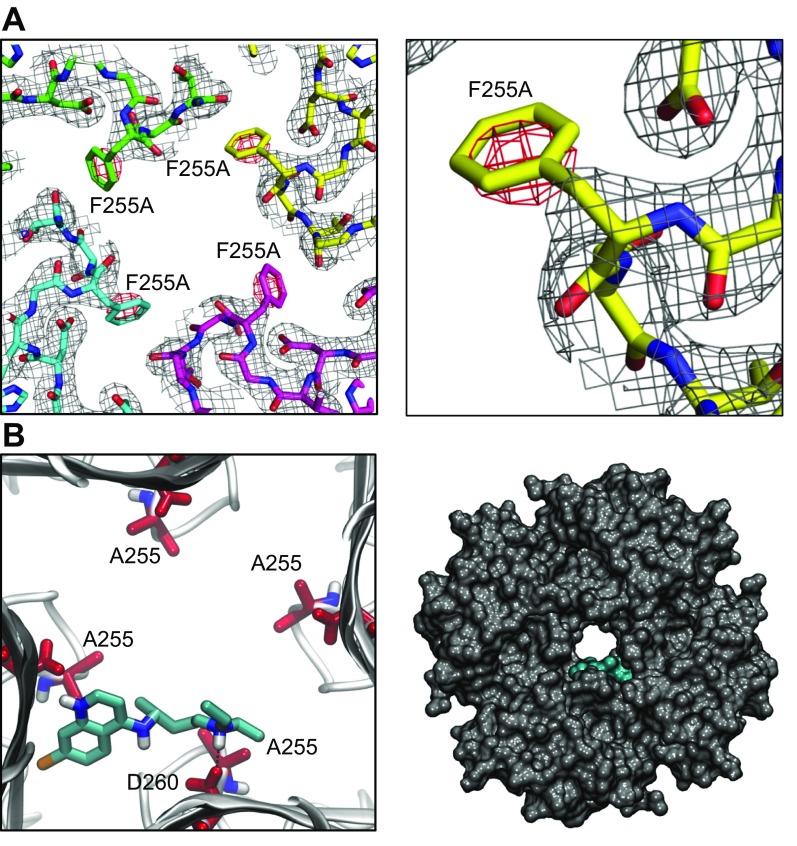
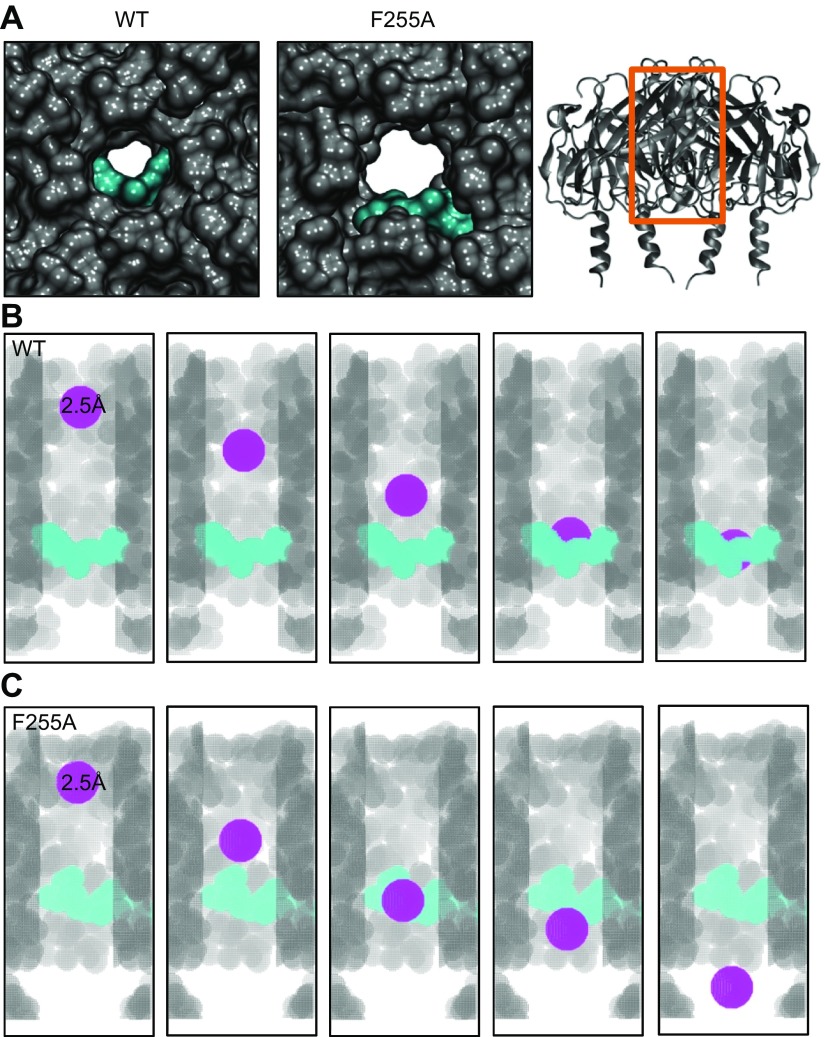
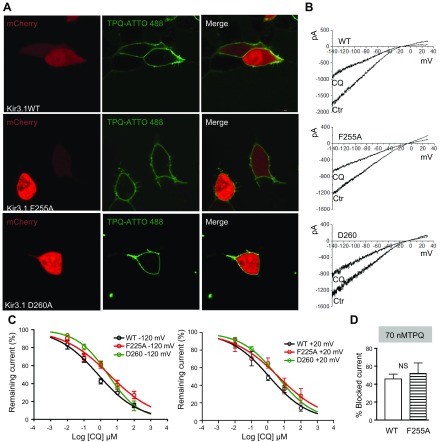
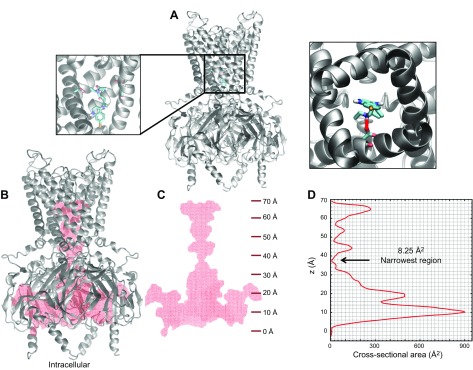
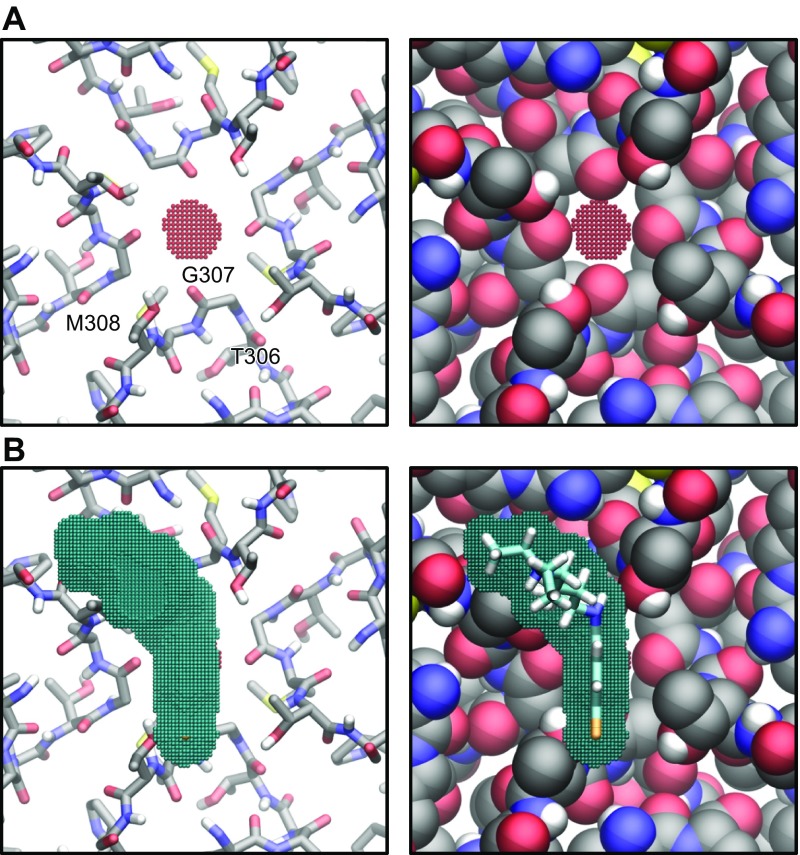
The acetylcholine-activated inward rectifier potassium current ( IKACh) is constitutively active in persistent atrial fibrillation (AF). We tested the hypothesis that the blocking of IKACh with the small molecule chloroquine terminates persistent AF. We used a sheep model of tachypacing-induced, persistent AF, molecular modeling, electrophysiology, and structural biology approaches. The 50% inhibition/inhibitory concentration of IKACh block with chloroquine, measured by patch clamp, was 1 μM. In optical mapping of sheep hearts with persistent AF, 1 μM chloroquine restored sinus rhythm. Molecular modeling suggested that chloroquine blocked the passage of a hydrated potassium ion through the intracellular domain of Kir3.1 (a molecular correlate of IKACh) by interacting with residues D260 and F255, in proximity to I228, Q227, and L299. 1H 15N heteronuclear single-quantum correlation of purified Kir3.1 intracellular domain confirmed the modeling results. F255, I228, Q227, and L299 underwent significant chemical-shift perturbations upon drug binding. We then crystallized and solved a 2.5 Å X-ray structure of Kir3.1 with F255A mutation. Modeling of chloroquine binding to the mutant channel suggested that the drug's binding to the pore becomes off centered, reducing its ability to block a hydrated potassium ion. Patch clamp validated the structural and modeling data, where the F255A and D260A mutations significantly reduced IKACh block by chloroquine. With the use of numerical and structural biology approaches, we elucidated the details of how a small molecule could block an ion channel and exert antiarrhythmic effects. Chloroquine binds the IKACh channel at a site formed by specific amino acids in the ion-permeation pathway, leading to decreased IKACh and the subsequent termination of AF.-Takemoto, Y., Slough, D. P., Meinke, G., Katnik, C., Graziano, Z. A., Chidipi, B., Reiser, M., Alhadidy, M. M., Ramirez, R., Salvador-Montañés, O., Ennis, S., Guerrero-Serna, G., Haburcak, M., Diehl, C., Cuevas, J., Jalife, J., Bohm, A., Lin,Y.-S., Noujaim, S. F. Structural basis for the antiarrhythmic blockade of a potassium channel with a small molecule.
Keywords: IKACh; atrial fibrillation; potassium inward rectifier.
Conflict of interest statement
The authors thank Dr. Kevin Nash (University of South Florida) for his help with creation of mutant constructs. This work was supported, in part, by an American Heart Association postdoctoral fellowship to Y.T.; U.S. National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (NHLBI), R21 HL138064 and R01HL129136 to S.F.N.; Tufts start-up and the Knez Family Investment Funds for Y-S.L.; and NIH NHLBI R01HL122352, the Leducq Foundation: Transatlantic Network of Excellence Program on Structural Alterations in the Myocardium and the Substrate for Cardiac Fibrillation, and University of Michigan Health System–Peking University Health Science Center Joint Institute for Translational and Clinical Research to J.J. HEK293 cells, stably transfected with Kir3.1 and Kir3.4, were a kind gift from the laboratory of Dr. Douglas Bayliss (University of Virginia Charlottesville, VA, USA). The authors declare no conflicts of interest.
Figures
References
-
- Makary S., Voigt N., Maguy A., Wakili R., Nishida K., Harada M., Dobrev D., Nattel S. (2011) Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine-regulated potassium channels in atrial remodeling. Circ. Res. 109, 1031–1043 10.1161/CIRCRESAHA.111.253120 - DOI - PubMed
-
- Voigt N., Trausch A., Knaut M., Matschke K., Varró A., Van Wagoner D. R., Nattel S., Ravens U., Dobrev D. (2010) Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circ Arrhythm Electrophysiol 3, 472–480 10.1161/CIRCEP.110.954636 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
