

Late-Onset Cryopyrin-Associated Periodic Syndromes Caused by Somatic NLRP3 Mosaicism-UK Single Center Experience
- PMID: 29163488
- PMCID: PMC5671490
- DOI: 10.3389/fimmu.2017.01410
Late-Onset Cryopyrin-Associated Periodic Syndromes Caused by Somatic NLRP3 Mosaicism-UK Single Center Experience
Abstract
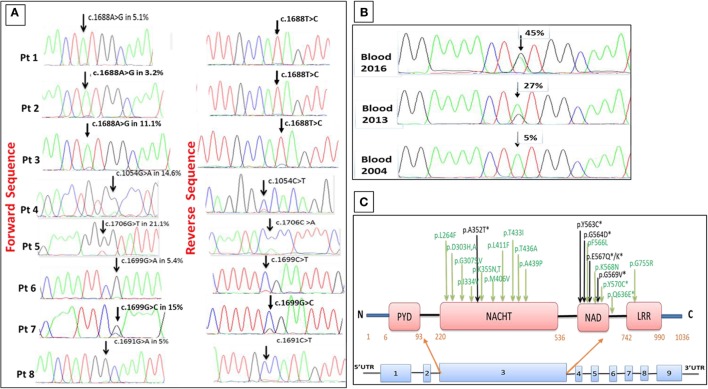
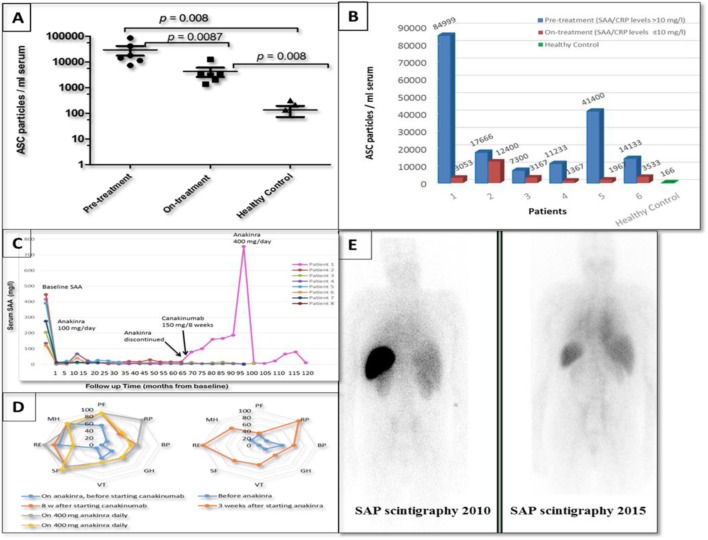
Cryopyrin-associated periodic syndrome (CAPS) is caused by gain-of-function NLRP3 mutations. Recently, somatic NLRP3 mosaicism has been reported in some CAPS patients who were previously classified as "mutation-negative." We describe here the clinical and laboratory findings in eight British adult patients who presented with symptoms typical of CAPS other than an onset in mid-late adulthood. All patients underwent comprehensive clinical and laboratory investigations, including analysis of the NLRP3 gene using Sanger and amplicon-based deep sequencing (ADS) along with measurements of extracellular apoptosis-associated speck-like protein with CARD domain (ASC) aggregates. The clinical phenotype in all subjects was consistent with mid-spectrum CAPS, except a median age at disease onset of 50 years. Sanger sequencing of NLRP3 was non-diagnostic but ADS detected a somatic NLRP3 mutation in each case. In one patient, DNA isolated from blood demonstrated an increase in the mutant allele from 5 to 45% over 12 years. ASC aggregates in patients' serum measured during active disease were significantly higher than healthy controls. This series represents 8% of CAPS patients diagnosed in a single center, suggesting that acquired NLRP3 mutations may not be an uncommon cause of the syndrome and should be sought in all patients with late-onset symptoms otherwise compatible with CAPS. Steadily worsening CAPS symptoms in one patient were associated with clonal expansion of the mutant allele predominantly affecting myeloid cells. Two patients developed AA amyloidosis, which previously has only been reported in CAPS in association with life-long germline NLRP3 mutations.
Keywords: AA amyloidosis; ASC aggregates; IL-1β; NLRP3 somatic mutation; cryopyrin-associated periodic syndrome; mutant allele.
Figures
References
-
- Kile RL, Rusk HA. A case of cold urticaria with an unusual family history. JAMA (1940) 114:1067–8.
-
- Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum (2002) 46:3340–8.10.1002/art.10688 - DOI - PMC - PubMed
-
- Feldmann J, Prieur A-M, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet (2002) 71:198–203.10.1086/341357 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous