

Rapid, ultra low coverage copy number profiling of cell-free DNA as a precision oncology screening strategy
- PMID: 29163793
- PMCID: PMC5685714
- DOI: 10.18632/oncotarget.21163
Rapid, ultra low coverage copy number profiling of cell-free DNA as a precision oncology screening strategy
Abstract
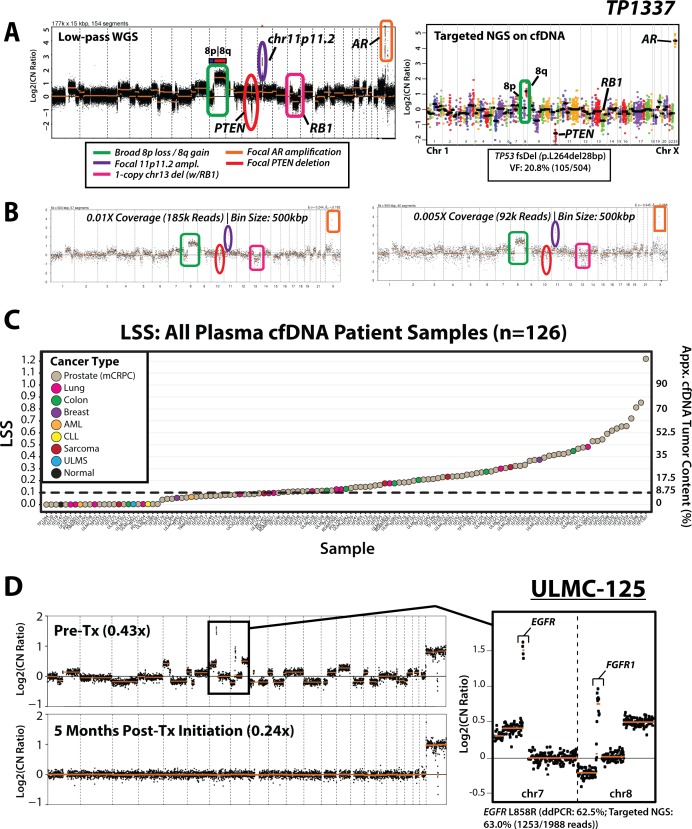
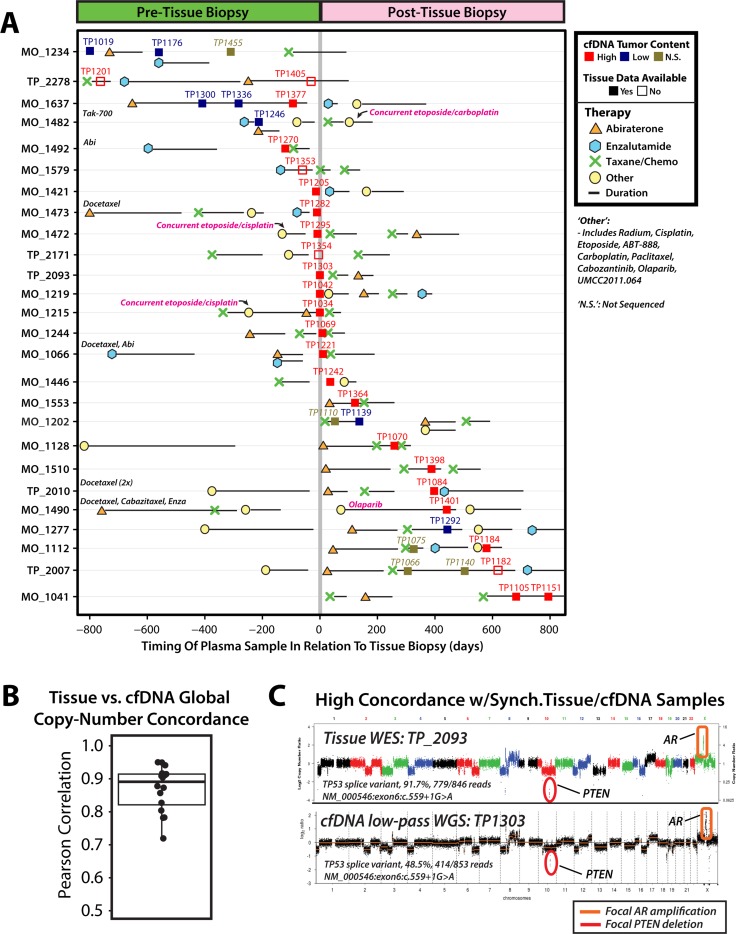
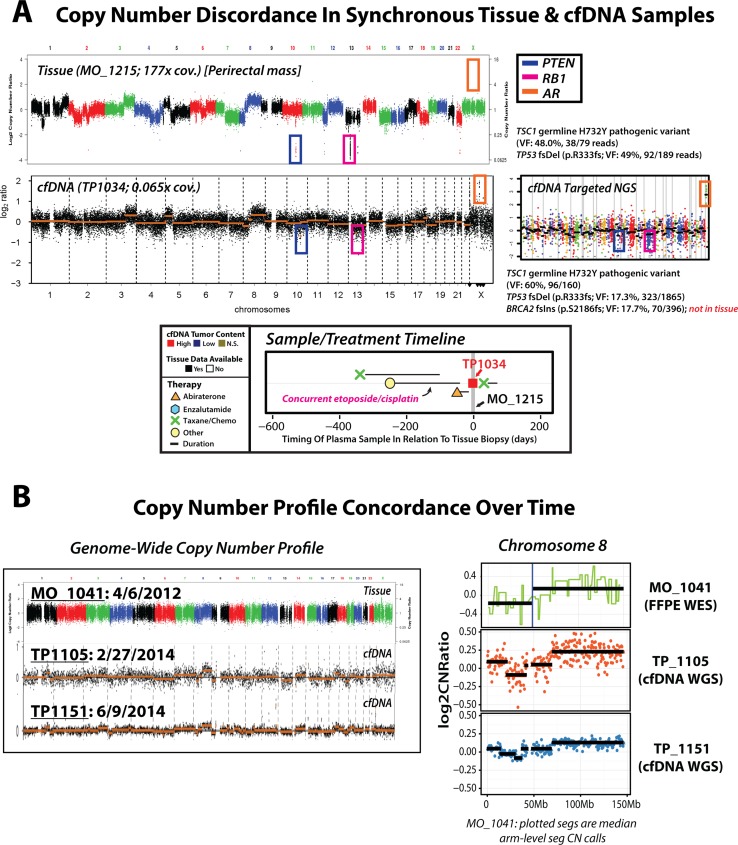
Current cell-free DNA (cfDNA) next generation sequencing (NGS) precision oncology workflows are typically limited to targeted and/or disease-specific applications. In advanced cancer, disease burden and cfDNA tumor content are often elevated, yielding unique precision oncology opportunities. We sought to demonstrate the utility of a pan-cancer, rapid, inexpensive, whole genome NGS of cfDNA approach (PRINCe) as a precision oncology screening strategy via ultra-low coverage (~0.01x) tumor content determination through genome-wide copy number alteration (CNA) profiling. We applied PRINCe to a retrospective cohort of 124 cfDNA samples from 100 patients with advanced cancers, including 76 men with metastatic castration-resistant prostate cancer (mCRPC), enabling cfDNA tumor content approximation and actionable focal CNA detection, while facilitating concordance analyses between cfDNA and tissue-based NGS profiles and assessment of cfDNA alteration associations with mCRPC treatment outcomes. Therapeutically relevant focal CNAs were present in 42 (34%) cfDNA samples, including 36 of 93 (39%) mCRPC patient samples harboring AR amplification. PRINCe identified pre-treatment cfDNA CNA profiles facilitating disease monitoring. Combining PRINCe with routine targeted NGS of cfDNA enabled mutation and CNA assessment with coverages tuned to cfDNA tumor content. In mCRPC, genome-wide PRINCe cfDNA and matched tissue CNA profiles showed high concordance (median Pearson correlation = 0.87), and PRINCe detectable AR amplifications predicted reduced time on therapy, independent of therapy type (Kaplan-Meier log-rank test, chi-square = 24.9, p < 0.0001). Our screening approach enables robust, broadly applicable cfDNA-based precision oncology for patients with advanced cancer through scalable identification of therapeutically relevant CNAs and pre-/post-treatment genomic profiles, enabling cfDNA- or tissue-based precision oncology workflow optimization.
Keywords: cell-free DNA; copy-number analysis; precision oncology; prostate cancer; whole genome sequencing.
Conflict of interest statement
CONFLICTS OF INTEREST C.H., T.T., J.L. and E.K. are current or former employees of Takara Bio USA. D.H.H. has received travel support from Thermo Fisher. S.A.T has received travel support from, and had a sponsored research agreement with Compendia Bioscience/Life Technologies/ThermoFisher that provided access to the targeted sequencing panel used herein. No other aspect of this study was supported by Compendia Bioscience/Life Technologies/ThermoFisher. The University of Michigan has been issued a patent on ETS gene fusions in prostate cancer on which A.M.C. and S.A.T. are co-inventors. The diagnostic field of use has been licensed to Hologic/Gen-Probe, Inc., which has sublicensed rights to Roche/Ventana Medical Systems. S.A.T. has an unrelated sponsored research agreement with Astellas. S.A.T. has served as a consultant for and received honoraria from Roche/Ventana Medical Systems, Almac Diagnostics, Janssen, AbbVie and Astellas/Medivation. S.A.T. is a co-founder of, consultant for and Laboratory Director of Strata Oncology. The other authors have no competing interests to declare.
Figures


References
-
- Roychowdhury S, Chinnaiyan AM. Translating cancer genomes and transcriptomes for precision oncology. CA Cancer J Clin. 2016;66:75–88. https://doi.org/10.3322/caac.21329. - DOI - PMC - PubMed
-
- Hyman DM, Solit DB, Arcila ME, Cheng DT, Sabbatini P, Baselga J, Berger MF, Ladanyi M. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today. 2015;20:1422–8. https://doi.org/10.1016/j.drudis.2015.08.005. - DOI - PMC - PubMed
-
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, Brannon AR, O’Reilly C, Sadowska J, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–64. https://doi.org/10.1016/j.jmoldx.2014.12.006. - DOI - PMC - PubMed
-
- Hovelson DH, McDaniel AS, Cani AK, Johnson B, Rhodes K, Williams PD, Bandla S, Bien G, Choppa P, Hyland F, Gottimukkala R, Liu G, Manivannan M, et al. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia. 2015;17:385–99. https://doi.org/10.1016/j.neo.2015.03.004. - DOI - PMC - PubMed
-
- Grasso C, Butler T, Rhodes K, Quist M, Neff TL, Moore S, Tomlins SA, Reinig E, Beadling C, Andersen M, Corless CL. Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data. J Mol Diagn. 2015;17:53–63. https://doi.org/10.1016/j.jmoldx.2014.09.008. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
