

A Universal Approach to Correct Various HBB Gene Mutations in Human Stem Cells for Gene Therapy of Beta-Thalassemia and Sickle Cell Disease
- PMID: 29164808
- PMCID: PMC5746148
- DOI: 10.1002/sctm.17-0066
A Universal Approach to Correct Various HBB Gene Mutations in Human Stem Cells for Gene Therapy of Beta-Thalassemia and Sickle Cell Disease
Abstract
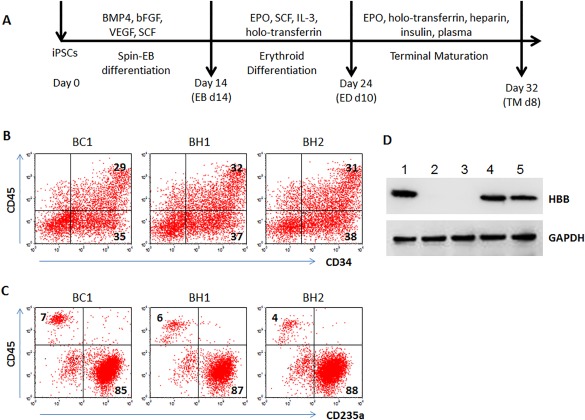
Beta-thalassemia is one of the most common recessive genetic diseases, caused by mutations in the HBB gene. Over 200 different types of mutations in the HBB gene containing three exons have been identified in patients with β-thalassemia (β-thal) whereas a homozygous mutation in exon 1 causes sickle cell disease (SCD). Novel therapeutic strategies to permanently correct the HBB mutation in stem cells that are able to expand and differentiate into erythrocytes producing corrected HBB proteins are highly desirable. Genome editing aided by CRISPR/Cas9 and other site-specific engineered nucleases offers promise to precisely correct a genetic mutation in the native genome without alterations in other parts of the human genome. Although making a sequence-specific nuclease to enhance correction of a specific HBB mutation by homology-directed repair (HDR) is becoming straightforward, targeting various HBB mutations of β-thal is still challenging because individual guide RNA as well as a donor DNA template for HDR of each type of HBB gene mutation have to be selected and validated. Using human induced pluripotent stem cells (iPSCs) from two β-thal patients with different HBB gene mutations, we devised and tested a universal strategy to achieve targeted insertion of the HBB cDNA in exon 1 of HBB gene using Cas9 and two validated guide RNAs. We observed that HBB protein production was restored in erythrocytes derived from iPSCs of two patients. This strategy of restoring functional HBB gene expression will be able to correct most types of HBB gene mutations in β-thal and SCD. Stem Cells Translational Medicine 2018;7:87-97.
Keywords: Beta-thalassemia; CRISPR/Cas9; Gene editing; Gene therapy; Hemoglobinopathies; Stem cells.
© 2017 The Authors Stem Cells Translational Medicine published by Wiley Periodicals, Inc. on behalf of AlphaMed Press.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
