

Identification of diverse target RNAs that are functionally regulated by human Pumilio proteins
- PMID: 29165587
- PMCID: PMC5758885
- DOI: 10.1093/nar/gkx1120
Identification of diverse target RNAs that are functionally regulated by human Pumilio proteins
Abstract
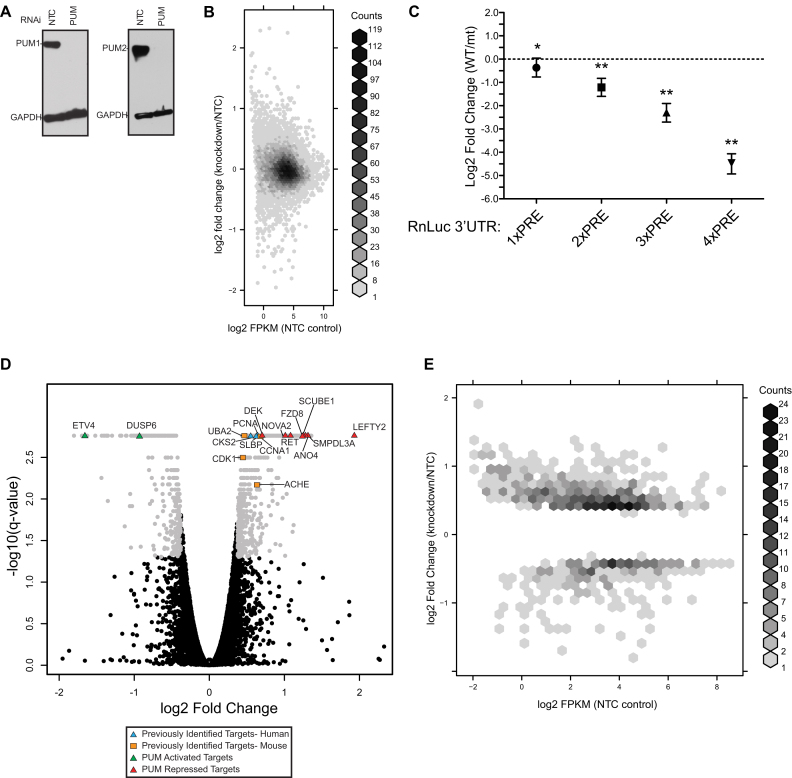
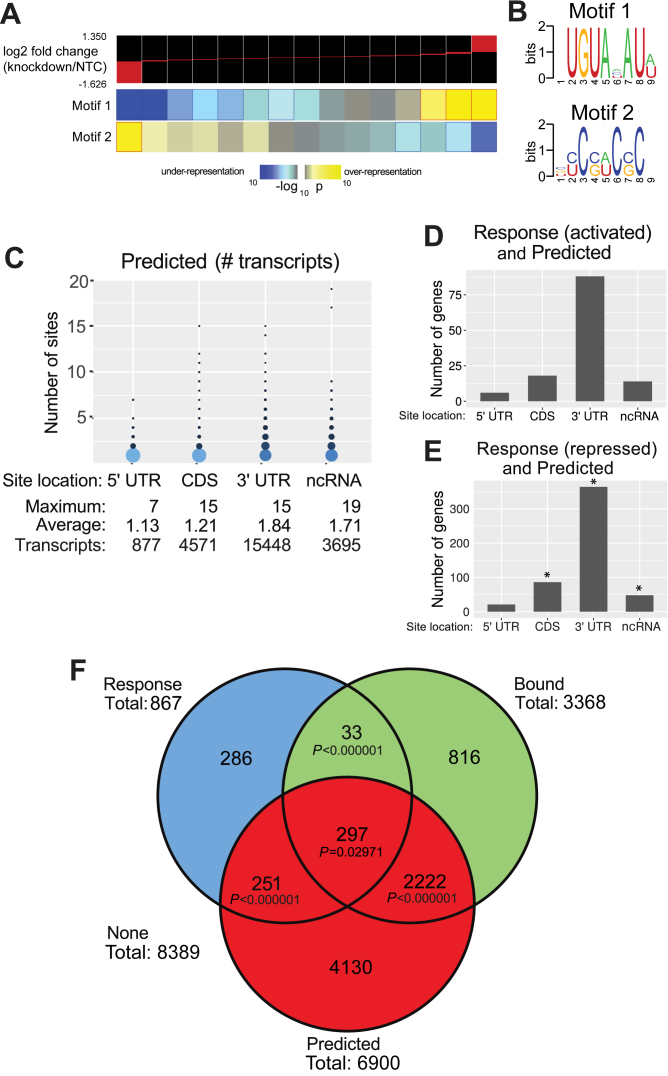
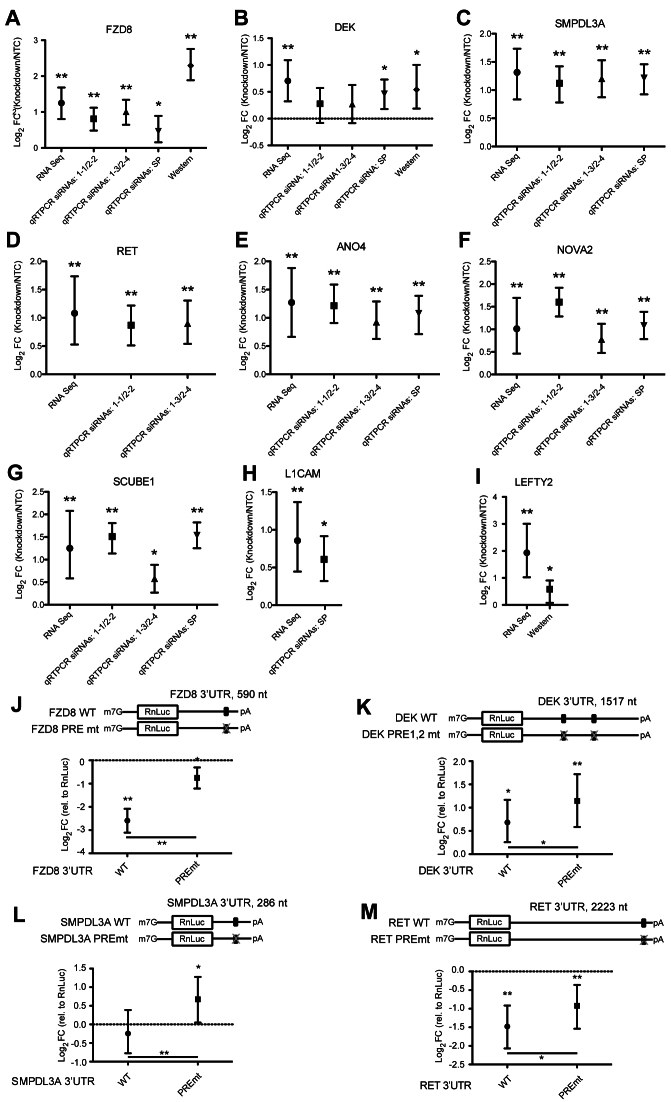
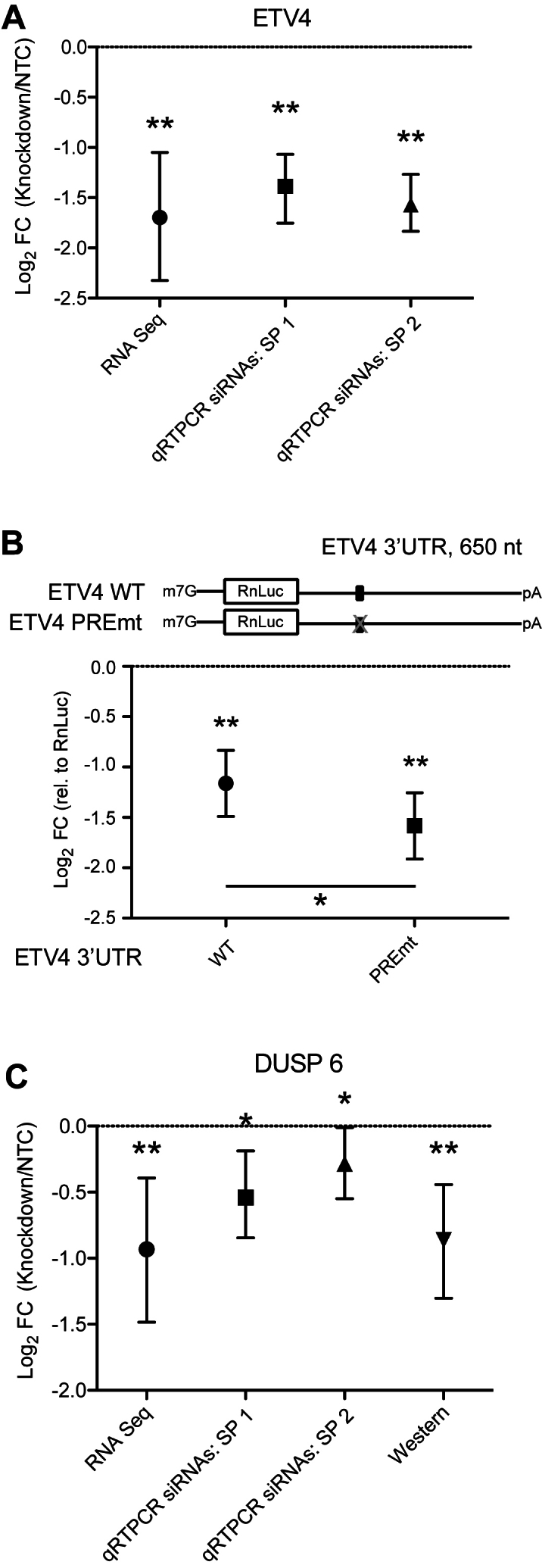
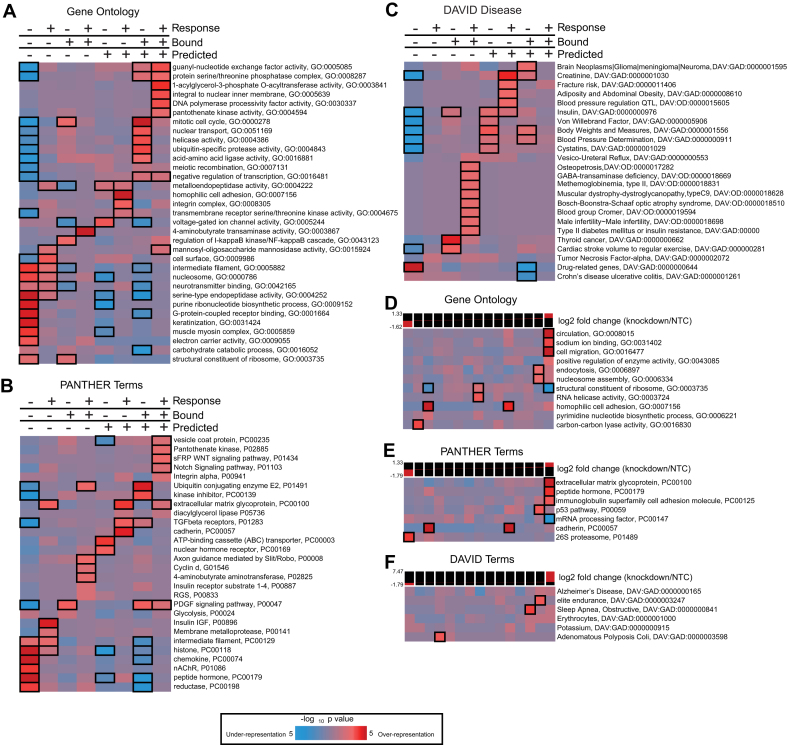
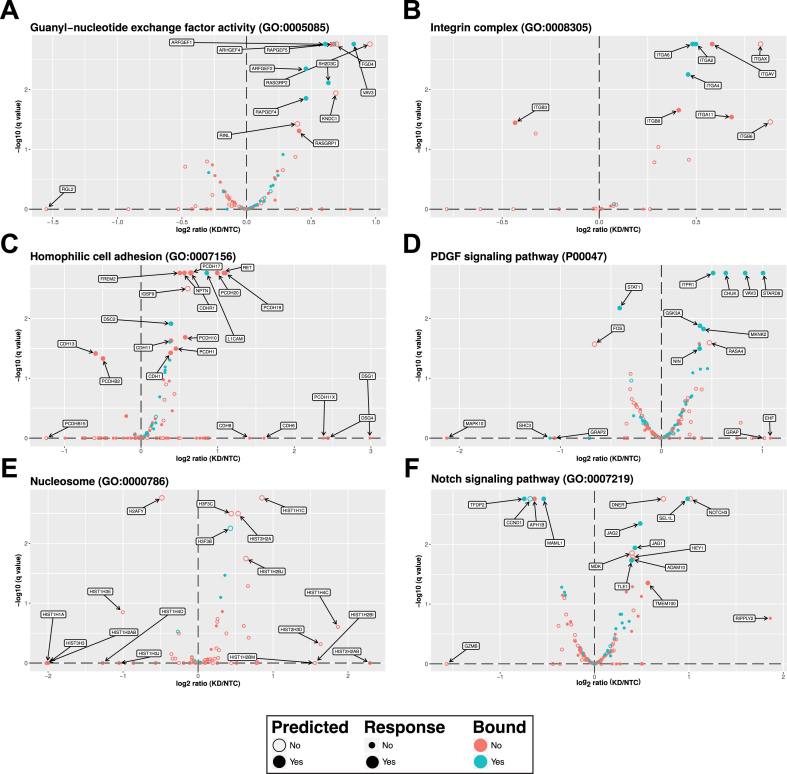
Human Pumilio proteins, PUM1 and PUM2, are sequence specific RNA-binding proteins that regulate protein expression. We used RNA-seq, rigorous statistical testing and an experimentally derived fold change cut-off to identify nearly 1000 target RNAs-including mRNAs and non-coding RNAs-that are functionally regulated by PUMs. Bioinformatic analysis defined a PUM Response Element (PRE) that was significantly enriched in transcripts that increased in abundance and matches the PUM RNA-binding consensus. We created a computational model that incorporates PRE position and frequency within an RNA relative to the magnitude of regulation. The model reveals significant correlation of PUM regulation with PREs in 3' untranslated regions (UTRs), coding sequences and non-coding RNAs, but not 5' UTRs. To define direct, high confidence PUM targets, we cross-referenced PUM-regulated RNAs with all PRE-containing RNAs and experimentally defined PUM-bound RNAs. The results define nearly 300 direct targets that include both PUM-repressed and, surprisingly, PUM-activated target RNAs. Annotation enrichment analysis reveal that PUMs regulate genes from multiple signaling pathways and developmental and neurological processes. Moreover, PUM target mRNAs impinge on human disease genes linked to cancer, neurological disorders and cardiovascular disease. These discoveries pave the way for determining how the PUM-dependent regulatory network impacts biological functions and disease states.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Antagonistic and cooperative AGO2-PUM interactions in regulating mRNAs.Sci Rep. 2018 Oct 17;8(1):15316. doi: 10.1038/s41598-018-33596-4. Sci Rep. 2018. PMID: 30333515 Free PMC article.
-
PUM1 and PUM2 exhibit different modes of regulation for SIAH1 that involve cooperativity with NANOS paralogues.Cell Mol Life Sci. 2019 Jan;76(1):147-161. doi: 10.1007/s00018-018-2926-5. Epub 2018 Sep 29. Cell Mol Life Sci. 2019. PMID: 30269240 Free PMC article.
-
Principles of mRNA control by human PUM proteins elucidated from multimodal experiments and integrative data analysis.RNA. 2020 Nov;26(11):1680-1703. doi: 10.1261/rna.077362.120. Epub 2020 Aug 4. RNA. 2020. PMID: 32753408 Free PMC article.
-
The Role of Pumilio RNA Binding Protein in Plants.Biomolecules. 2021 Dec 9;11(12):1851. doi: 10.3390/biom11121851. Biomolecules. 2021. PMID: 34944494 Free PMC article. Review.
-
Post-transcriptional Regulatory Functions of Mammalian Pumilio Proteins.Trends Genet. 2018 Dec;34(12):972-990. doi: 10.1016/j.tig.2018.09.006. Epub 2018 Oct 10. Trends Genet. 2018. PMID: 30316580 Free PMC article. Review.
Cited by
-
Regulation of Parkinson's disease-associated genes by Pumilio proteins and microRNAs in SH-SY5Y neuronal cells.PLoS One. 2022 Sep 29;17(9):e0275235. doi: 10.1371/journal.pone.0275235. eCollection 2022. PLoS One. 2022. PMID: 36174040 Free PMC article.
-
A Quantitative and Predictive Model for RNA Binding by Human Pumilio Proteins.Mol Cell. 2019 Jun 6;74(5):966-981.e18. doi: 10.1016/j.molcel.2019.04.012. Epub 2019 May 8. Mol Cell. 2019. PMID: 31078383 Free PMC article.
-
RNA molecular recording with an engineered RNA deaminase.Nat Methods. 2023 Dec;20(12):1887-1899. doi: 10.1038/s41592-023-02046-z. Epub 2023 Oct 19. Nat Methods. 2023. PMID: 37857907 Free PMC article.
-
Core spliceosomal Sm proteins as constituents of cytoplasmic mRNPs in plants.Plant J. 2020 Aug;103(3):1155-1173. doi: 10.1111/tpj.14792. Epub 2020 May 28. Plant J. 2020. PMID: 32369637 Free PMC article.
-
AGO-RBP crosstalk on target mRNAs: Implications in miRNA-guided gene silencing and cancer.Transl Oncol. 2022 Jul;21:101434. doi: 10.1016/j.tranon.2022.101434. Epub 2022 Apr 26. Transl Oncol. 2022. PMID: 35477066 Free PMC article. Review.
References
-
- Rajewsky N. microRNA target predictions in animals. Nat. Genet. 2006; 38(Suppl):S8–S13. - PubMed
-
- Wickens M., Bernstein D.S., Kimble J., Parker R. A PUF family portrait: 3΄UTR regulation as a way of life. Trends Genet. 2002; 18:150–157. - PubMed
-
- Zhang B., Gallegos M., Puoti A., Durkin E., Fields S., Kimble J., Wickens M.P. A conserved RNA-binding protein that regulates sexual fates in the C. elegans hermaphrodite germ line. Nature. 1997; 390:477–484. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
