

Targeting BCR-ABL-Independent TKI Resistance in Chronic Myeloid Leukemia by mTOR and Autophagy Inhibition
- PMID: 29165716
- PMCID: PMC5946859
- DOI: 10.1093/jnci/djx236
Targeting BCR-ABL-Independent TKI Resistance in Chronic Myeloid Leukemia by mTOR and Autophagy Inhibition
Abstract
Background: Imatinib and second-generation tyrosine kinase inhibitors (TKIs) nilotinib and dasatinib have statistically significantly improved the life expectancy of chronic myeloid leukemia (CML) patients; however, resistance to TKIs remains a major clinical challenge. Although ponatinib, a third-generation TKI, improves outcomes for patients with BCR-ABL-dependent mechanisms of resistance, including the T315I mutation, a proportion of patients may have or develop BCR-ABL-independent resistance and fail ponatinib treatment. By modeling ponatinib resistance and testing samples from these CML patients, it is hoped that an alternative drug target can be identified and inhibited with a novel compound.
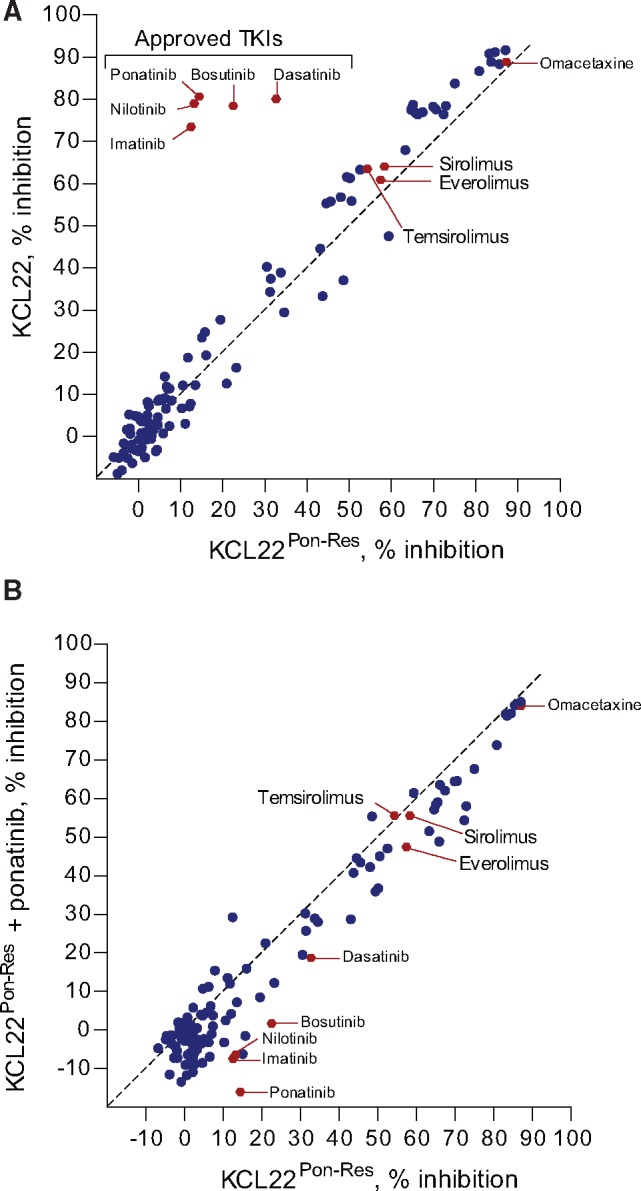
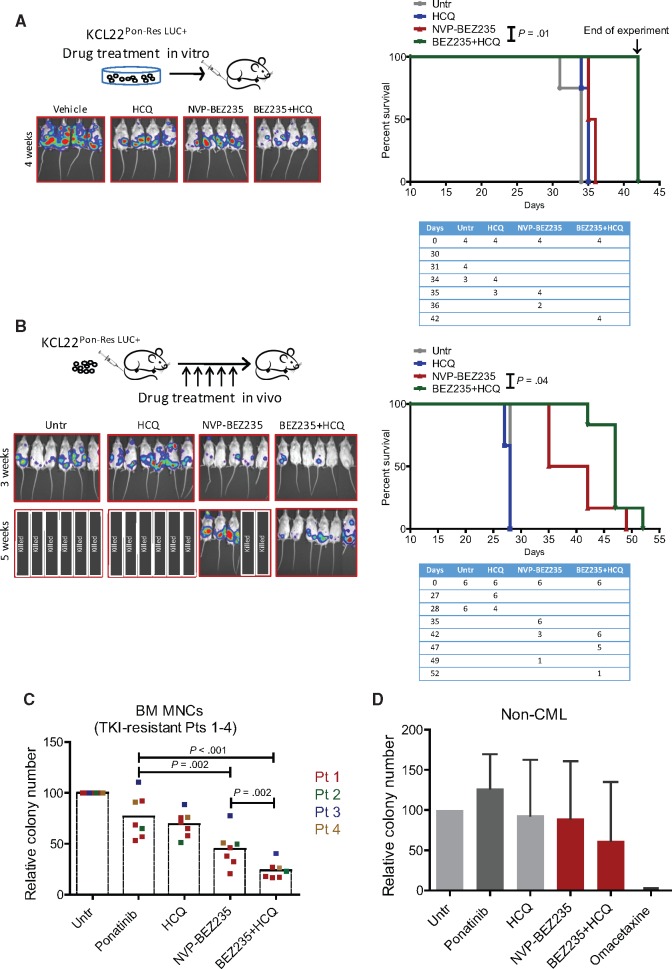
Methods: Two CML cell lines with acquired BCR-ABL-independent resistance were generated following culture in ponatinib. RNA sequencing and gene ontology (GO) enrichment were used to detect aberrant transcriptional response in ponatinib-resistant cells. A validated oncogene drug library was used to identify US Food and Drug Administration-approved drugs with activity against TKI-resistant cells. Validation was performed using bone marrow (BM)-derived cells from TKI-resistant patients (n = 4) and a human xenograft mouse model (n = 4-6 mice per group). All statistical tests were two-sided.
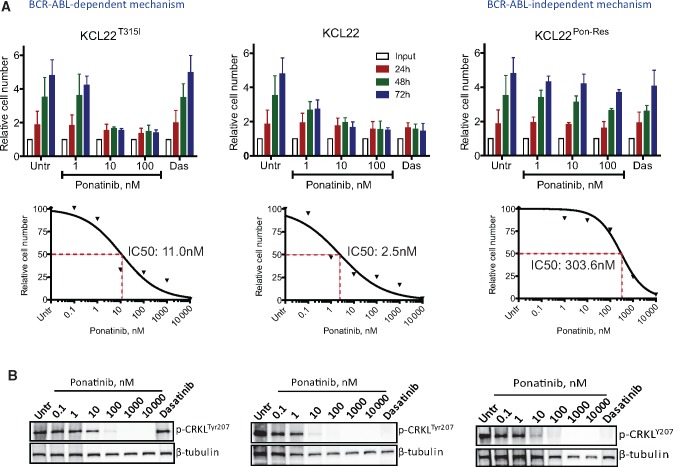
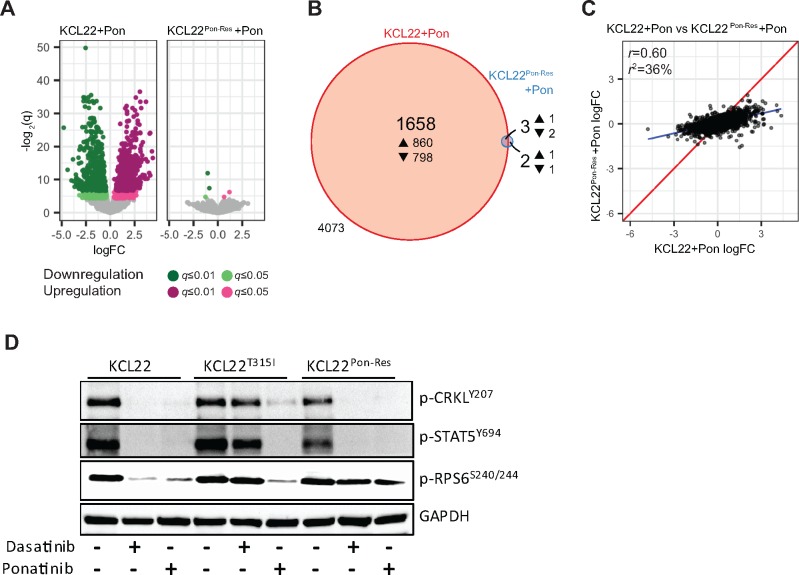
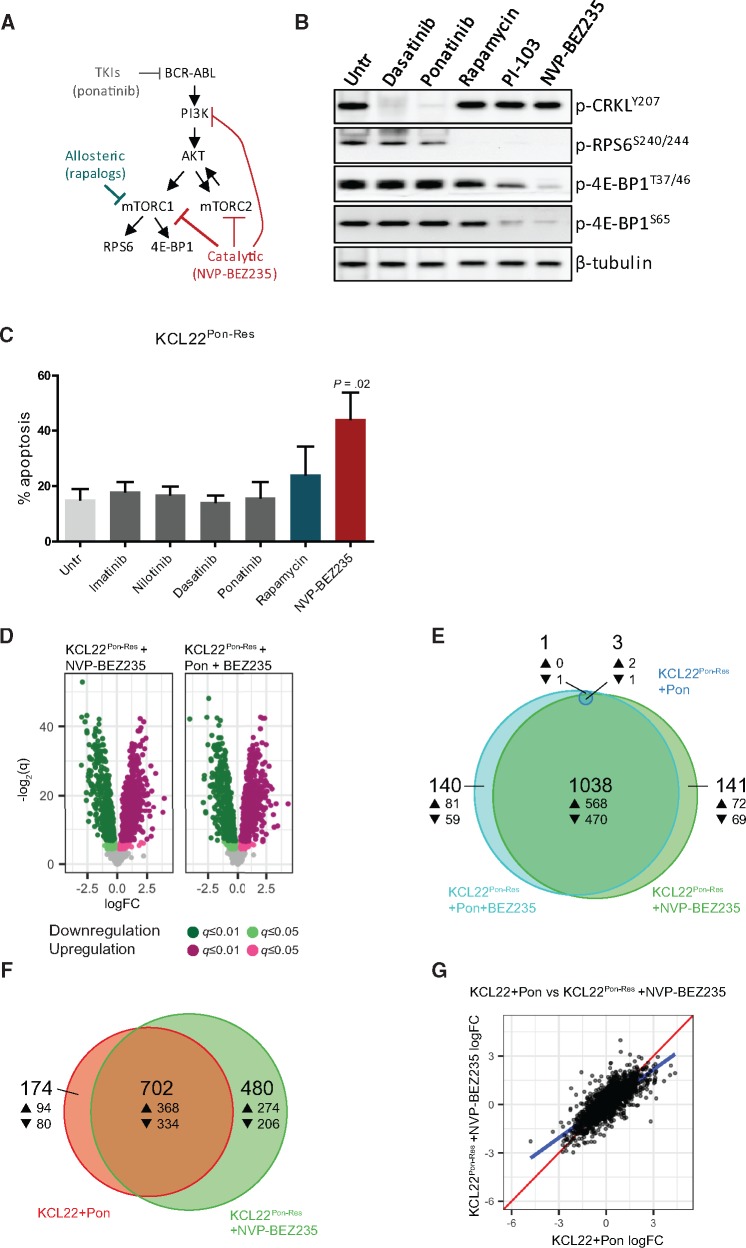
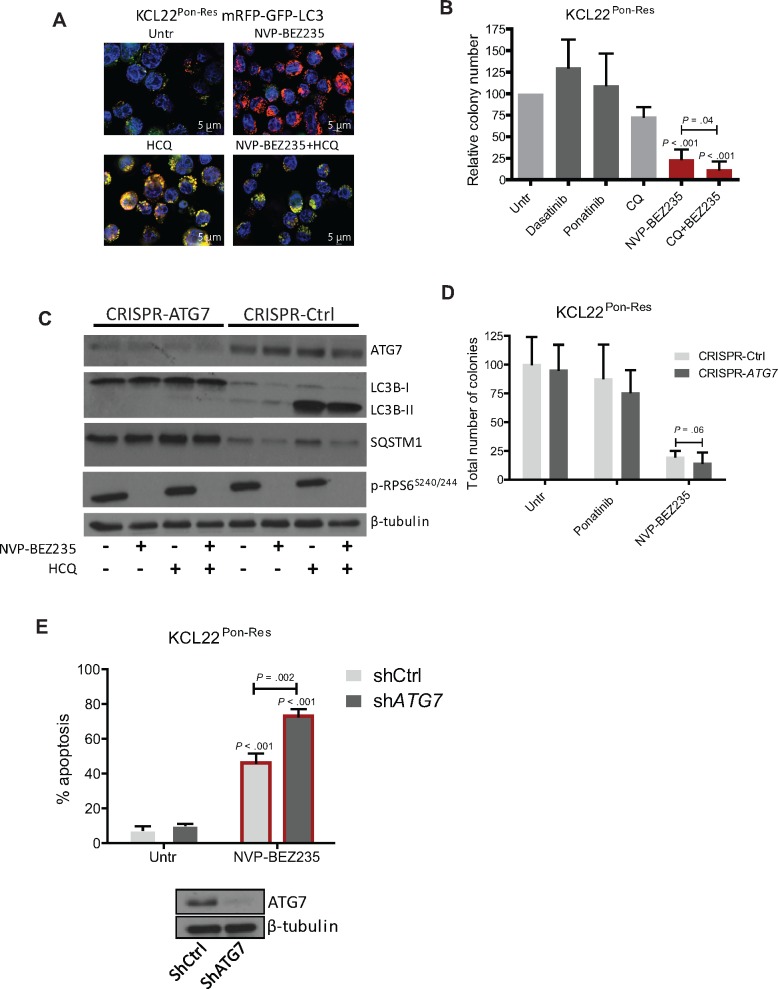
Results: We show that ponatinib-resistant CML cells can acquire BCR-ABL-independent resistance mediated through alternative activation of mTOR. Following transcriptomic analysis and drug screening, we highlight mTOR inhibition as an alternative therapeutic approach in TKI-resistant CML cells. Additionally, we show that catalytic mTOR inhibitors induce autophagy and demonstrate that genetic or pharmacological inhibition of autophagy sensitizes ponatinib-resistant CML cells to death induced by mTOR inhibition in vitro (% number of colonies of control[SD], NVP-BEZ235 vs NVP-BEZ235+HCQ: 45.0[17.9]% vs 24.0[8.4]%, P = .002) and in vivo (median survival of NVP-BEZ235- vs NVP-BEZ235+HCQ-treated mice: 38.5 days vs 47.0 days, P = .04).
Conclusion: Combined mTOR and autophagy inhibition may provide an attractive approach to target BCR-ABL-independent mechanism of resistance.
Figures
References
-
- Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–293.http://dx.doi.org/10.1038/243290a0 - DOI - PubMed
-
- Daley GQ, Van Etten RA, Baltimore D.. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247(4944):824–830.http://dx.doi.org/10.1126/science.2406902 - DOI - PubMed
-
- Druker BJ, Guilhot F, O'Brien SG et al. , Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417.http://dx.doi.org/10.1056/NEJMoa062867 - DOI - PubMed
-
- Graham SM, Jorgensen HG, Allan E et al. , Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–325.http://dx.doi.org/10.1182/blood.V99.1.319 - DOI - PubMed
-
- Corbin AS, Agarwal A, Loriaux M et al. , Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121(1):396–409.http://dx.doi.org/10.1172/JCI35721 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
