

Long-Read Sequencing of Human Cytomegalovirus Transcriptome Reveals RNA Isoforms Carrying Distinct Coding Potentials
- PMID: 29167532
- PMCID: PMC5700075
- DOI: 10.1038/s41598-017-16262-z
Long-Read Sequencing of Human Cytomegalovirus Transcriptome Reveals RNA Isoforms Carrying Distinct Coding Potentials
Abstract
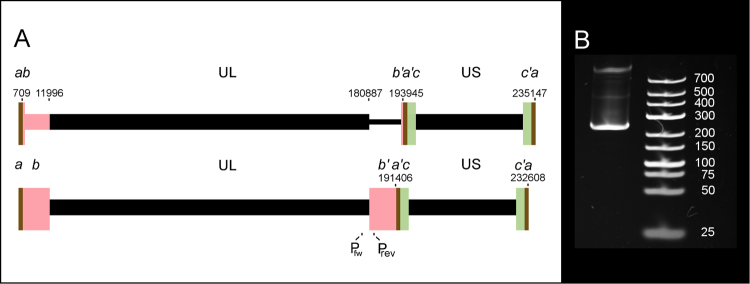
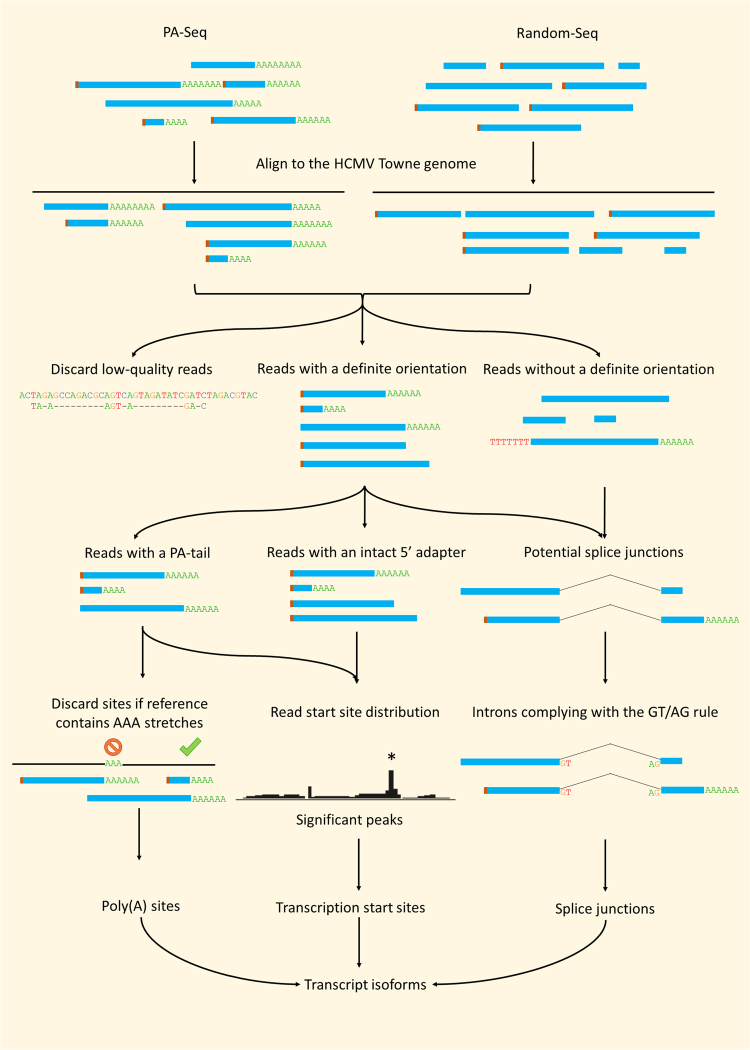
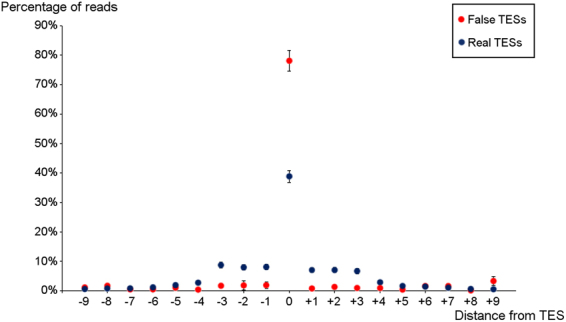
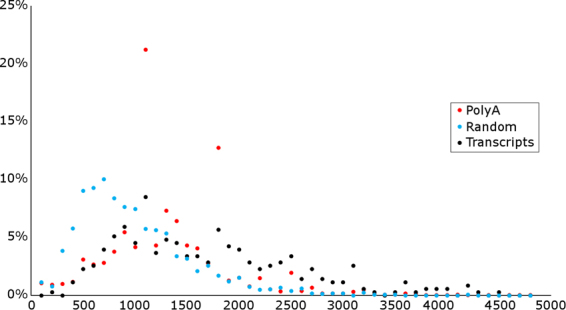
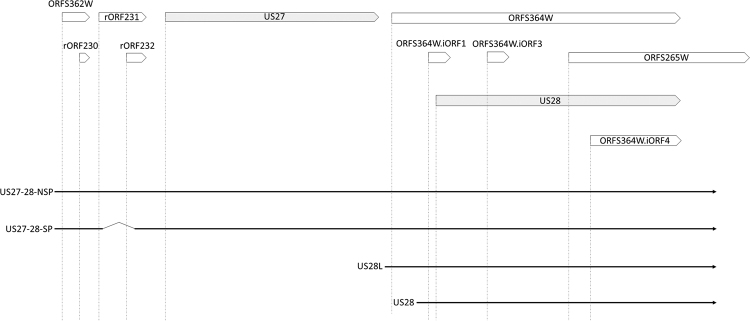
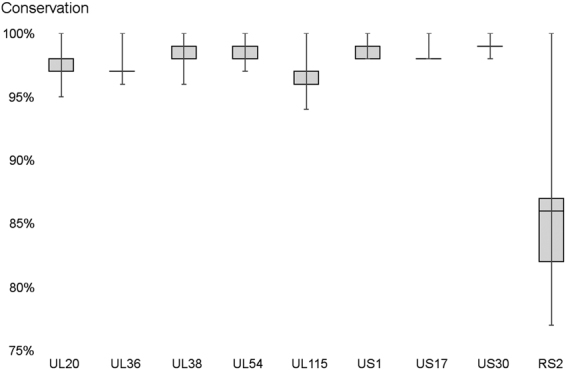
The human cytomegalovirus (HCMV) is a ubiquitous, human pathogenic herpesvirus. The complete viral genome is transcriptionally active during infection; however, a large part of its transcriptome has yet to be annotated. In this work, we applied the amplified isoform sequencing technique from Pacific Biosciences to characterize the lytic transcriptome of HCMV strain Towne varS. We developed a pipeline for transcript annotation using long-read sequencing data. We identified 248 transcriptional start sites, 116 transcriptional termination sites and 80 splicing events. Using this information, we have annotated 291 previously undescribed or only partially annotated transcript isoforms, including eight novel antisense transcripts and their isoforms, as well as a novel transcript (RS2) in the short repeat region, partially antisense to RS1. Similarly to other organisms, we discovered a high transcriptional diversity in HCMV, with many transcripts only slightly differing from one another. Comparing our transcriptome profiling results to an earlier ribosome footprint analysis, we have concluded that the majority of the transcripts contain multiple translationally active ORFs, and also that most isoforms contain unique combinations of ORFs. Based on these results, we propose that one important function of this transcriptional diversity may be to provide a regulatory mechanism at the level of translation.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
