

De novo design of a hyperstable non-natural protein-ligand complex with sub-Å accuracy
- PMID: 29168496
- PMCID: PMC5859929
- DOI: 10.1038/nchem.2846
De novo design of a hyperstable non-natural protein-ligand complex with sub-Å accuracy
Abstract
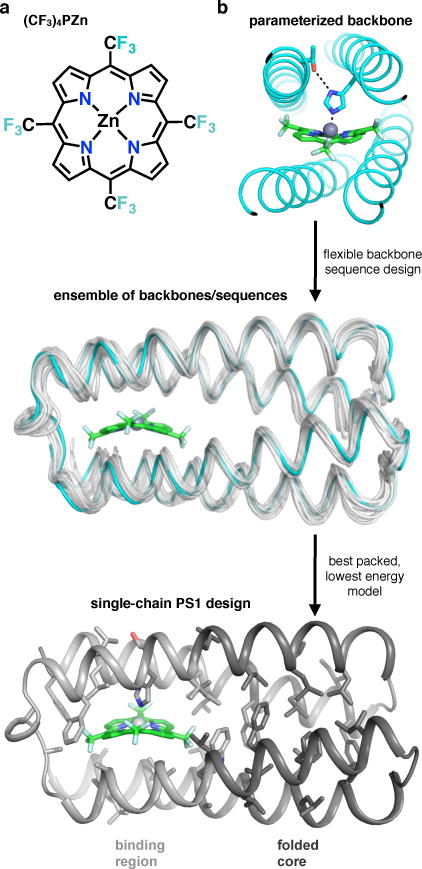
Protein catalysis requires the atomic-level orchestration of side chains, substrates and cofactors, and yet the ability to design a small-molecule-binding protein entirely from first principles with a precisely predetermined structure has not been demonstrated. Here we report the design of a novel protein, PS1, that binds a highly electron-deficient non-natural porphyrin at temperatures up to 100 °C. The high-resolution structure of holo-PS1 is in sub-Å agreement with the design. The structure of apo-PS1 retains the remote core packing of the holoprotein, with a flexible binding region that is predisposed to ligand binding with the desired geometry. Our results illustrate the unification of core packing and binding-site definition as a central principle of ligand-binding protein design.
Conflict of interest statement
The authors declare no competing financial interests.
Figures












References
-
- Roy S, et al. A Protein Designed by Binary Patterning of Polar and Nonpolar Amino Acids Displays Native-like Properties. J Am Chem Soc. 1997;119:5302–5306.
-
- Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. - PubMed
-
- Peacock AFA. Incorporating metals into de novo proteins. Curr Opin Chem Biol. 2013;17:934–939. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
