

DNA replication stress and cancer chemotherapy
- PMID: 29168596
- PMCID: PMC5797825
- DOI: 10.1111/cas.13455
DNA replication stress and cancer chemotherapy
Abstract
DNA replication is one of the fundamental biological processes in which dysregulation can cause genome instability. This instability is one of the hallmarks of cancer and confers genetic diversity during tumorigenesis. Numerous experimental and clinical studies have indicated that most tumors have experienced and overcome the stresses caused by the perturbation of DNA replication, which is also referred to as DNA replication stress (DRS). When we consider therapeutic approaches for tumors, it is important to exploit the differences in DRS between tumor and normal cells. In this review, we introduce the current understanding of DRS in tumors and discuss the underlying mechanism of cancer therapy from the aspect of DRS.
Keywords: DNA damage response; DNA replication stress; chemotherapeutic drugs; genome instability; tumorigenesis.
© 2017 The Authors. Cancer Science published by John Wiley & Sons Australia, Ltd on behalf of Japanese Cancer Association.
Figures
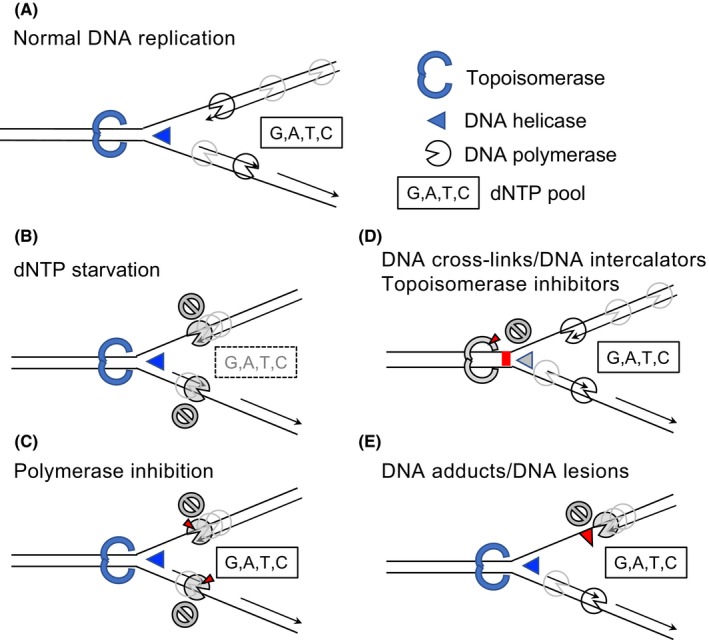
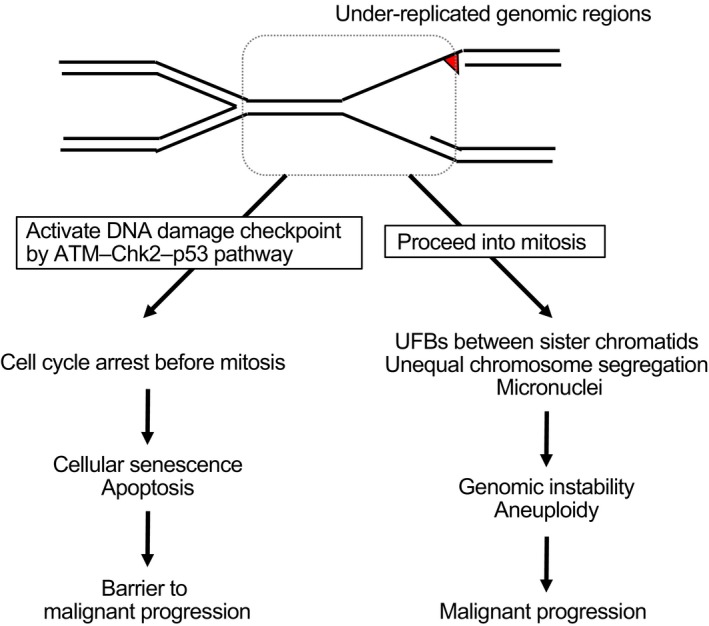
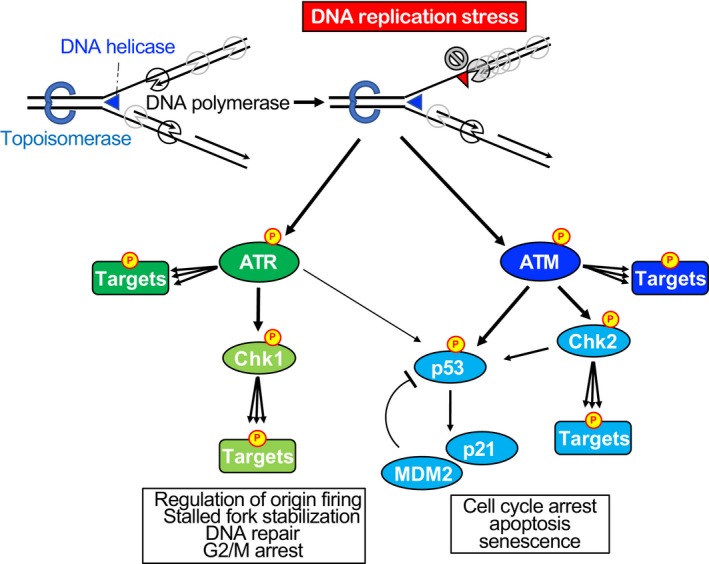
References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57‐70. - PubMed
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. - PubMed
-
- Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature. 2005;434:864‐870. - PubMed
-
- Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907‐913. - PubMed
-
- Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature. 2006;444:638‐642. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
