

Chemokines protect vascular smooth muscle cells from cell death induced by cyclic mechanical stretch
- PMID: 29170451
- PMCID: PMC5701048
- DOI: 10.1038/s41598-017-15867-8
Chemokines protect vascular smooth muscle cells from cell death induced by cyclic mechanical stretch
Abstract
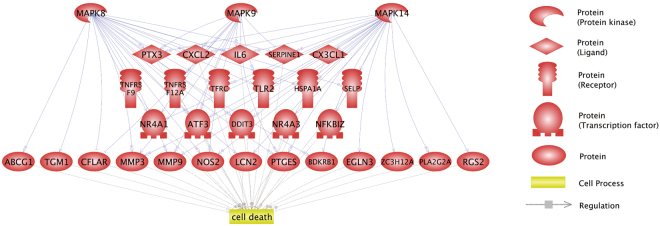
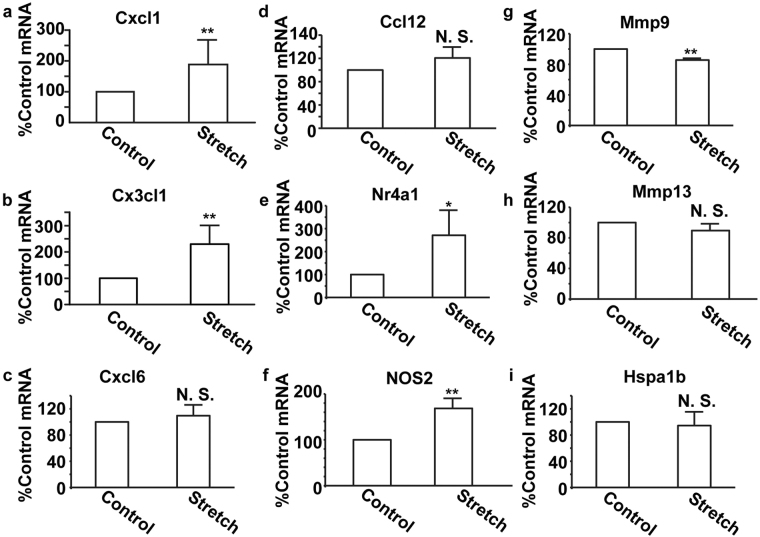
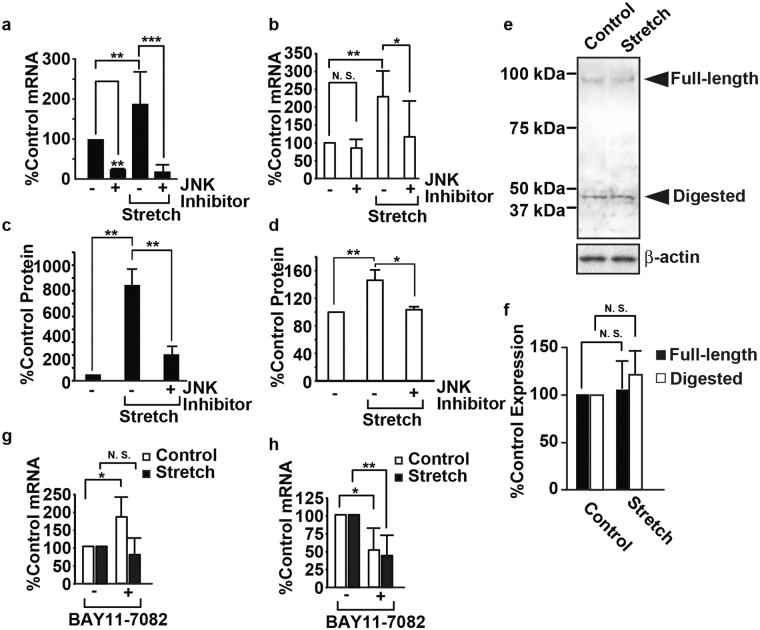
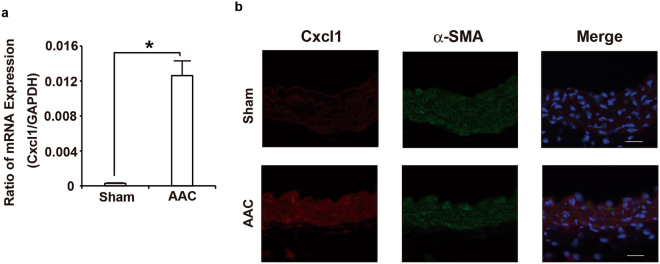
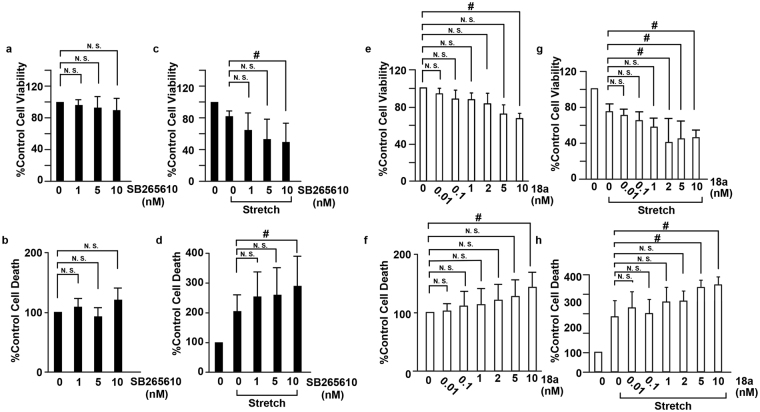
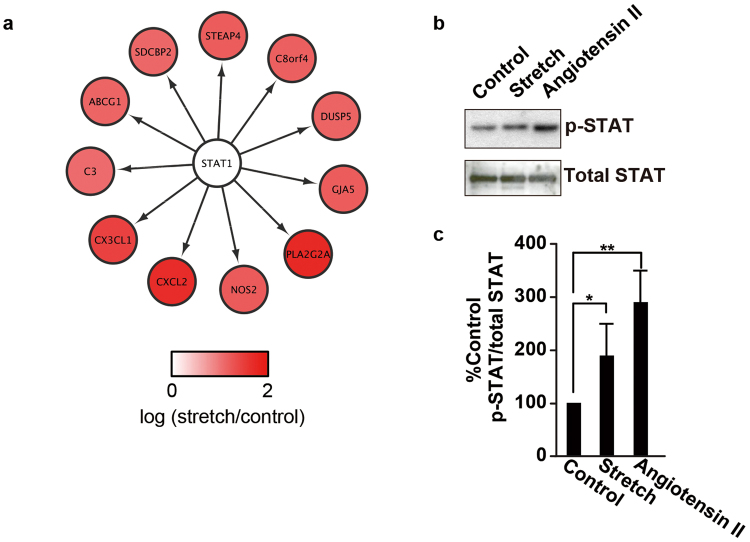
The pulsatile nature of blood flow exposes vascular smooth muscle cells (VSMCs) in the vessel wall to cyclic mechanical stretch (CMS), which evokes VSMC proliferation, cell death, phenotypic switching, and migration, leading to vascular remodeling. These responses have been observed in many cardiovascular diseases; however, the underlying mechanisms remain unclear. We have revealed that CMS of rat aortic smooth muscle cells (RASMCs) causes JNK- and p38-dependent cell death and that a calcium channel blocker and angiotensin II receptor antagonist decreased the phosphorylation of JNK and p38 and subsequently decreased cell death by CMS. In the present study, we showed that the expression of Cxcl1 and Cx3cl1 was induced by CMS in a JNK-dependent manner. The expression of Cxcl1 was also induced in VSMCs by hypertension produced by abdominal aortic constriction (AAC). In addition, antagonists against the receptors for CXCL1 and CX3CL1 increased cell death, indicating that CXCL1 and CX3CL1 protect RASMCs from CMS-induced cell death. We also revealed that STAT1 is activated in RASMCs subjected to CMS. Taken together, these results indicate that CMS of VSMCs induces inflammation-related gene expression, including that of CXCL1 and CX3CL1, which may play important roles in the stress response against CMS caused by hypertension.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Díez J, et al. Altered regulation of smooth muscle cell proliferation and apoptosis in small arteries of spontaneously hypertensive rats. Eur. Heart J. 1998;19:G29–33. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
