

Mosaicism in Cutaneous Disorders
- PMID: 29178821
- PMCID: PMC8026264
- DOI: 10.1146/annurev-genet-121415-121955
Mosaicism in Cutaneous Disorders
Abstract
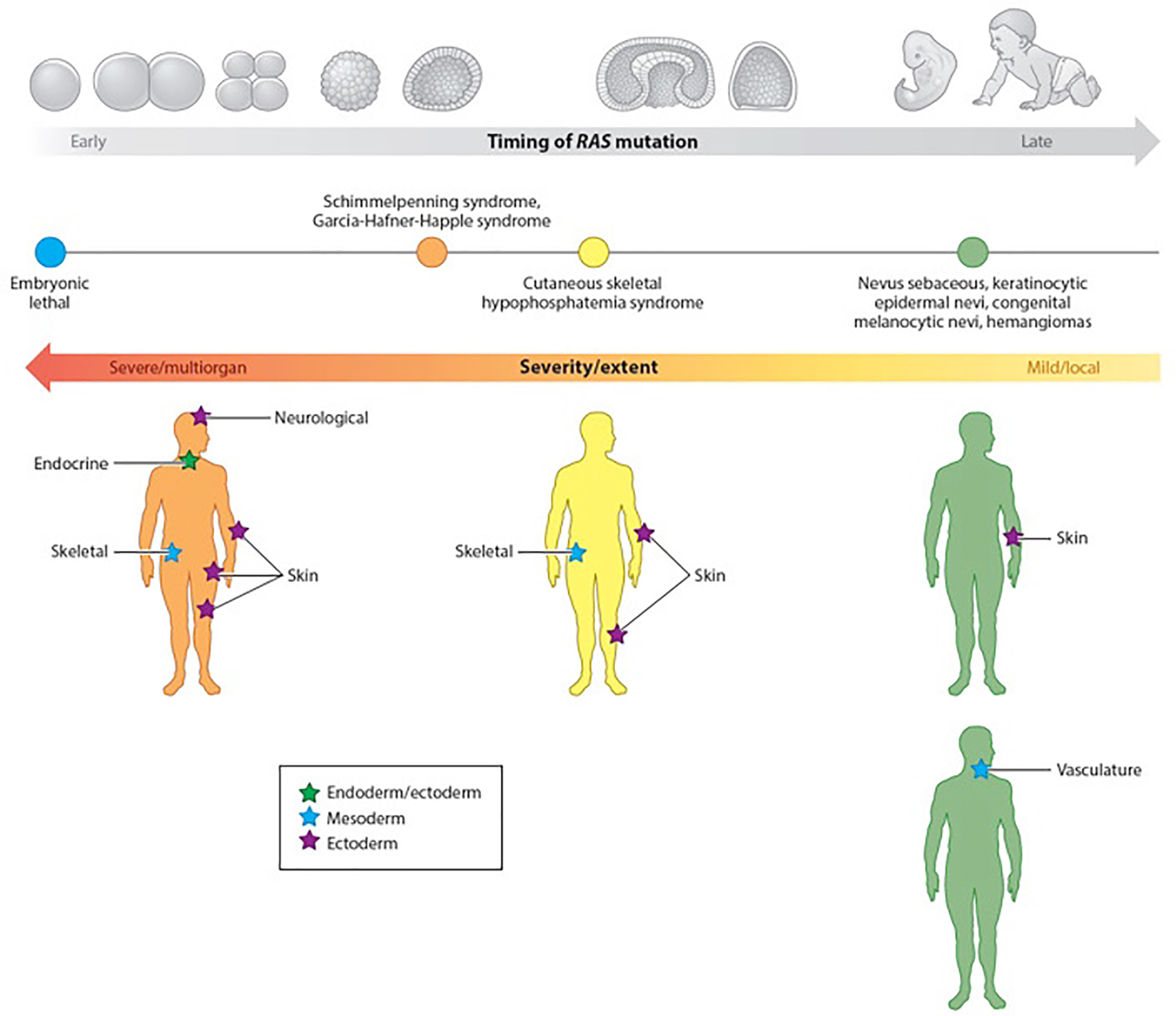
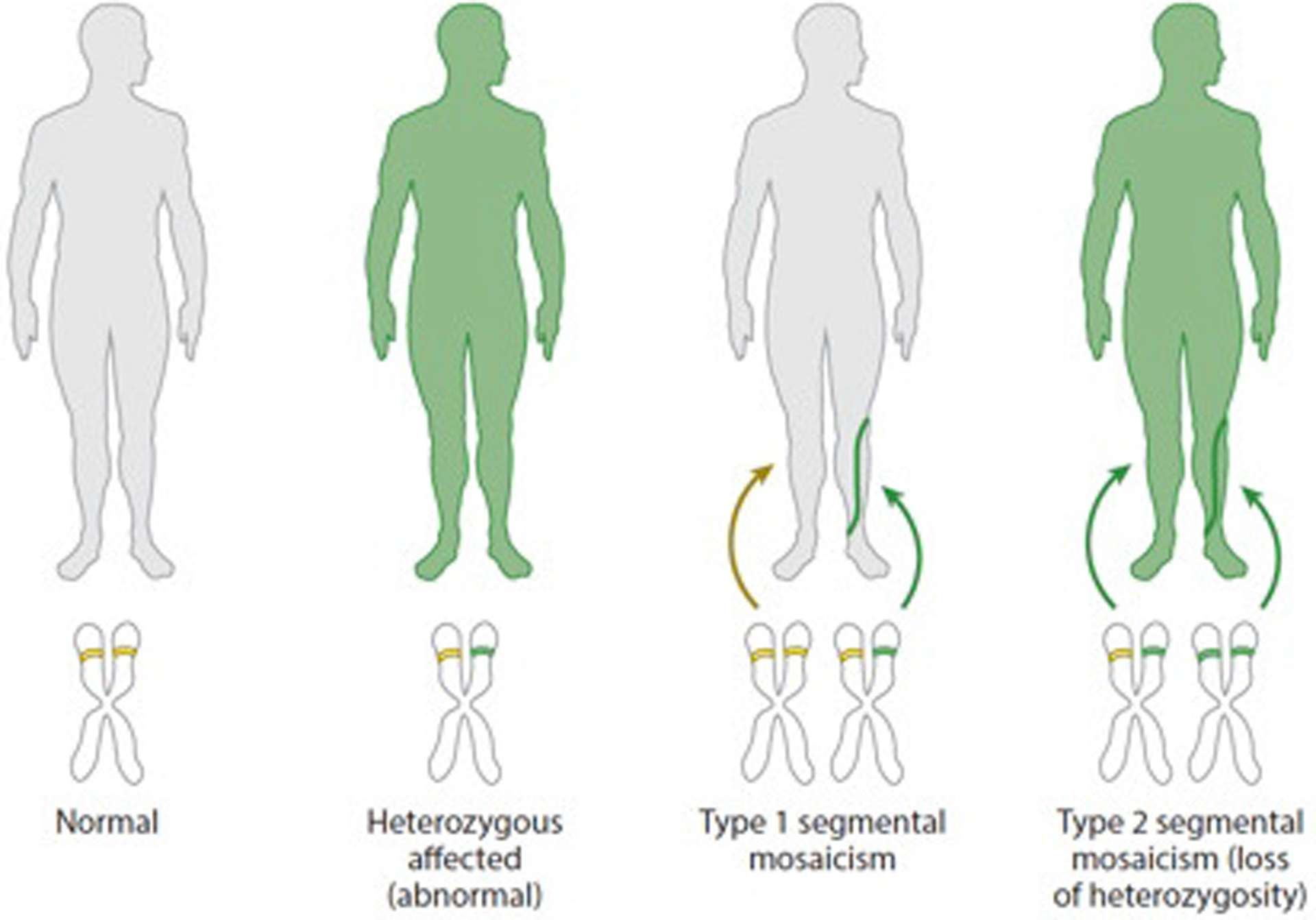
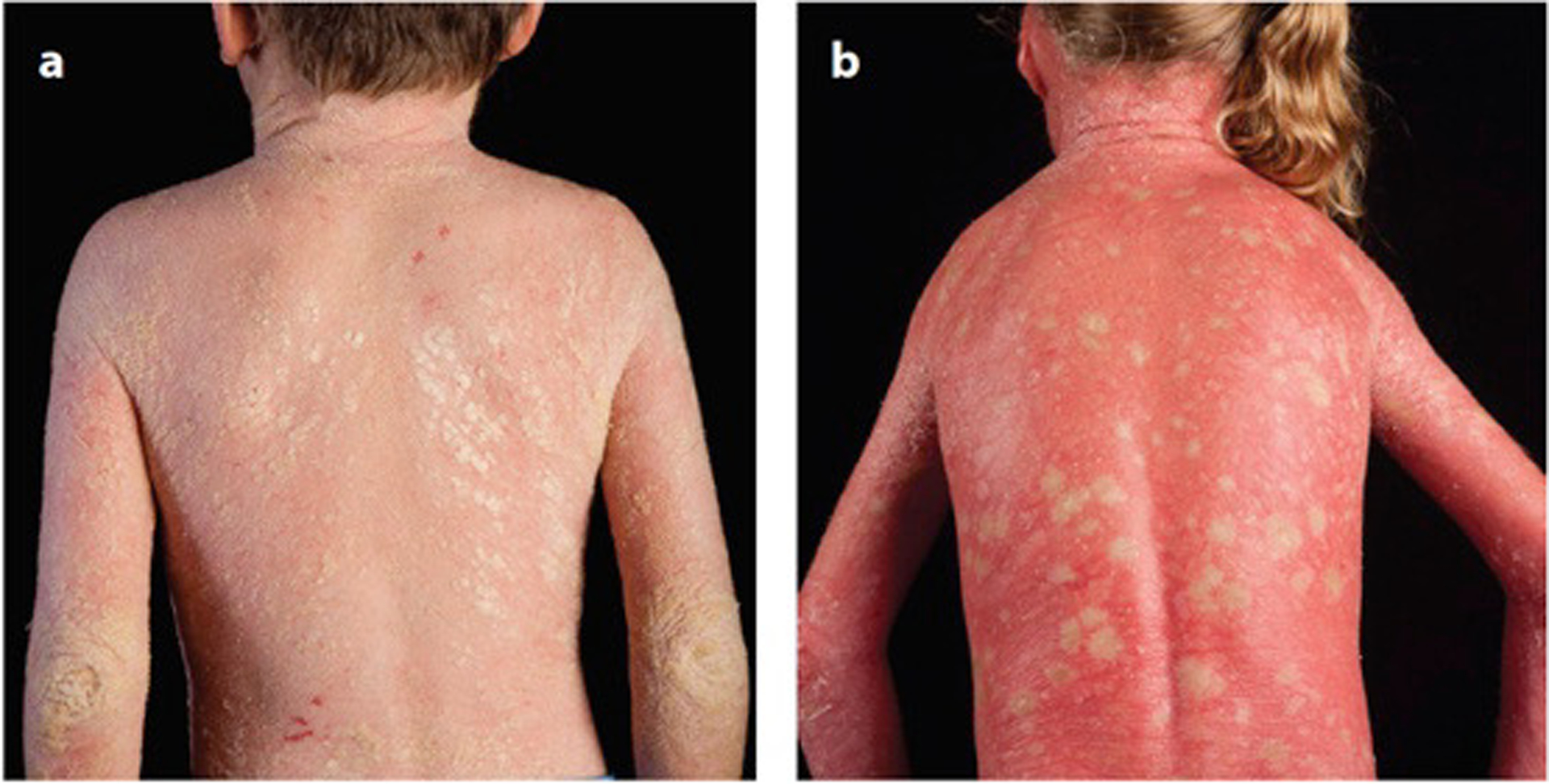
Genetic mosaicism arises when a zygote harbors two or more distinct genotypes, typically due to de novo, somatic mutation during embryogenesis. The clinical manifestations largely depend on the differentiation status of the mutated cell; earlier mutations target pluripotent cells and generate more widespread disease affecting multiple organ systems. If gonadal tissue is spared-as in somatic genomic mosaicism-the mutation and its effects are limited to the proband, whereas mosaicism also affecting the gametes, such as germline or gonosomal mosaicism, is transmissible. Mosaicism is easily appreciated in cutaneous disorders, as phenotypically distinct mutant cells often give rise to lesions in patterns determined by the affected cell type. Genetic investigation of cutaneous mosaic disorders has identified pathways central to disease pathogenesis, revealing novel therapeutic targets. In this review, we discuss examples of cutaneous mosaicism, approaches to gene discovery in these disorders, and insights into molecular pathobiology that have potential for clinical translation.
Keywords: RASopathy; epigenetic mosaicism; lines of Blaschko; nonsegmental mosaicism; revertant mosaicism; segmental mosaicism; somatic mutation.
Figures
References
-
- Ahuja YR. 1960. A human mosaic involving eye and hair color differences. Acta Genet. Med. Gemellol. (Roma) 9:427–31 - PubMed
-
- Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, et al. 2011. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat. Genet 43:1262–65 - PubMed
-
- Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, et al. 2005. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat. Genet 37:1038–40 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
