

Neurologic Phenotypes Associated With Mutations in RTN4IP1 (OPA10) in Children and Young Adults
- PMID: 29181510
- PMCID: PMC5833489
- DOI: 10.1001/jamaneurol.2017.2065
Neurologic Phenotypes Associated With Mutations in RTN4IP1 (OPA10) in Children and Young Adults
Abstract
Importance: Neurologic disorders with isolated symptoms or complex syndromes are relatively frequent among mitochondrial inherited diseases. Recessive RTN4IP1 gene mutations have been shown to cause isolated and syndromic optic neuropathies.
Objective: To define the spectrum of clinical phenotypes associated with mutations in RTN4IP1 encoding a mitochondrial quinone oxidoreductase.
Design, setting, and participants: This study involved 12 individuals from 11 families with severe central nervous system diseases and optic atrophy. Targeted and whole-exome sequencing were performed-at Hospital Angers (France), Institute of Neurology Milan (Italy), Imagine Institute Paris (France), Helmoltz Zentrum of Munich (Germany), and Beijing Genomics Institute (China)-to clarify the molecular diagnosis of patients. Each patient's neurologic, ophthalmologic, magnetic resonance imaging, and biochemical features were investigated. This study was conducted from May 1, 2014, to June 30, 2016.
Main outcomes and measures: Recessive mutations in RTN4IP1 were identified. Clinical presentations ranged from isolated optic atrophy to severe encephalopathies.
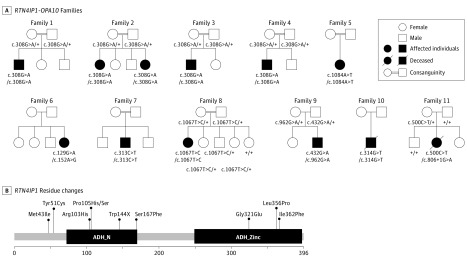
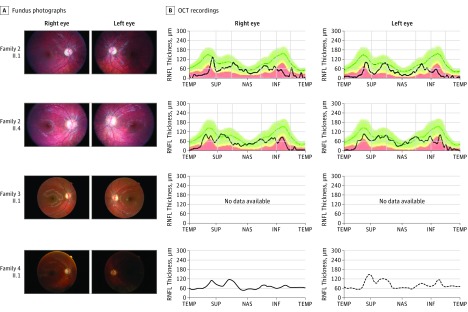
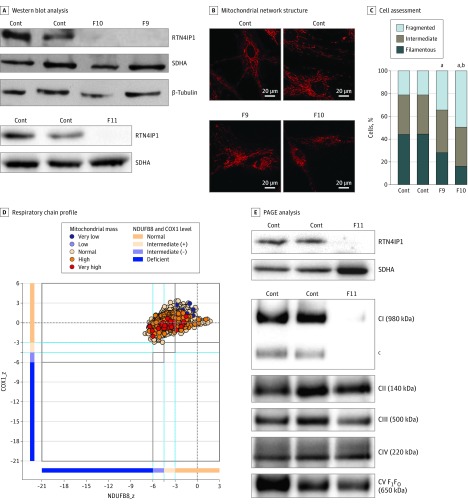
Results: Of the 12 individuals in the study, 6 (50%) were male and 6 (50%) were female. They ranged in age from 5 months to 32 years. Of the 11 families, 6 (5 of whom were consanguineous) had a member or members who presented isolated optic atrophy with the already reported p.Arg103His or the novel p.Ile362Phe, p.Met43Ile, and p.Tyr51Cys amino acid changes. The 5 other families had a member or members who presented severe neurologic syndromes with a common core of symptoms, including optic atrophy, seizure, intellectual disability, growth retardation, and elevated lactate levels. Additional clinical features of those affected were deafness, abnormalities on magnetic resonance images of the brain, stridor, and abnormal electroencephalographic patterns, all of which eventually led to death before age 3 years. In these patients, novel and very rare homozygous and compound heterozygous mutations were identified that led to the absence of the protein and complex I disassembly as well as mild mitochondrial network fragmentation.
Conclusions and relevance: A broad clinical spectrum of neurologic features, ranging from isolated optic atrophy to severe early-onset encephalopathies, is associated with RTN4IP1 biallelic mutations and should prompt RTN4IP1 screening in both syndromic neurologic presentations and nonsyndromic recessive optic neuropathies.
Conflict of interest statement
Figures
References
-
- DiMauro S. Mitochondrial diseases. Biochim Biophys Acta. 2004;1658(1-2):80-88. - PubMed
-
- Munnich A, Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am J Med Genet. 2001;106(1):4-17. - PubMed
-
- Hu WH, Hausmann ON, Yan MS, Walters WM, Wong PK, Bethea JR. Identification and characterization of a novel Nogo-interacting mitochondrial protein (NIMP). J Neurochem. 2002;81(1):36-45. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
