

Oxidative stress and cellular pathologies in Parkinson's disease
- PMID: 29183391
- PMCID: PMC5706368
- DOI: 10.1186/s13041-017-0340-9
Oxidative stress and cellular pathologies in Parkinson's disease
Abstract
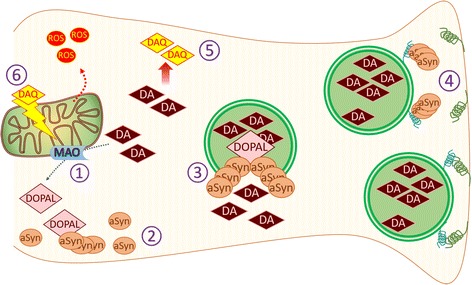
Parkinson's disease (PD) is a chronic and progressive neurodegeneration of dopamine neurons in the substantia nigra. The reason for the death of these neurons is unclear; however, studies have demonstrated the potential involvement of mitochondria, endoplasmic reticulum, α-synuclein or dopamine levels in contributing to cellular oxidative stress as well as PD symptoms. Even though those papers had separately described the individual roles of each element leading to neurodegeneration, recent publications suggest that neurodegeneration is the product of various cellular interactions. This review discusses the role of oxidative stress in mediating separate pathological events that together, ultimately result in cell death in PD. Understanding the multi-faceted relationships between these events, with oxidative stress as a common denominator underlying these processes, is needed for developing better therapeutic strategies.
Keywords: Alpha-synuclein; Dopamine neurons; Mitochondria; Oxidative stress; Parkinson’s disease; Reactive oxygen species; Unfolded protein response.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
