

Andrographolide Protects against Aortic Banding-Induced Experimental Cardiac Hypertrophy by Inhibiting MAPKs Signaling
- PMID: 29184496
- PMCID: PMC5694538
- DOI: 10.3389/fphar.2017.00808
Andrographolide Protects against Aortic Banding-Induced Experimental Cardiac Hypertrophy by Inhibiting MAPKs Signaling
Abstract
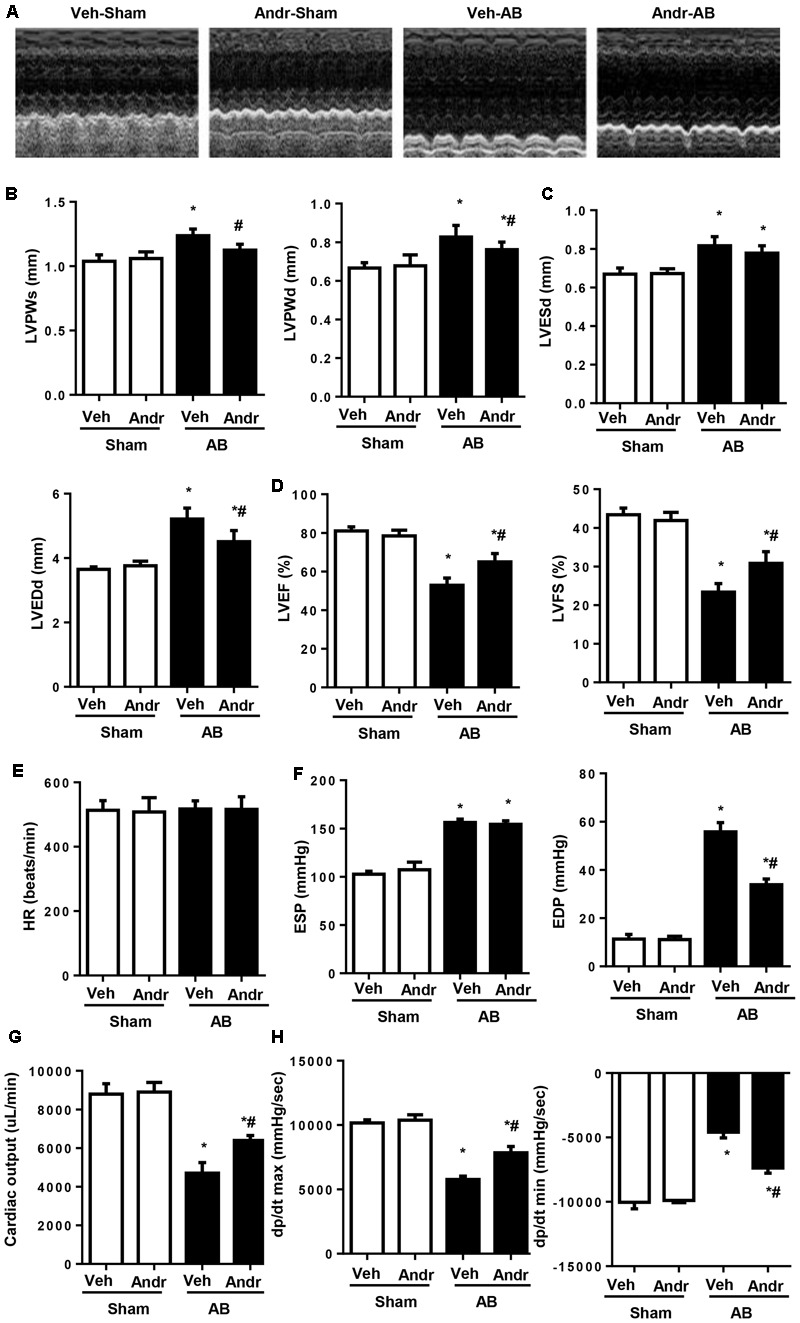
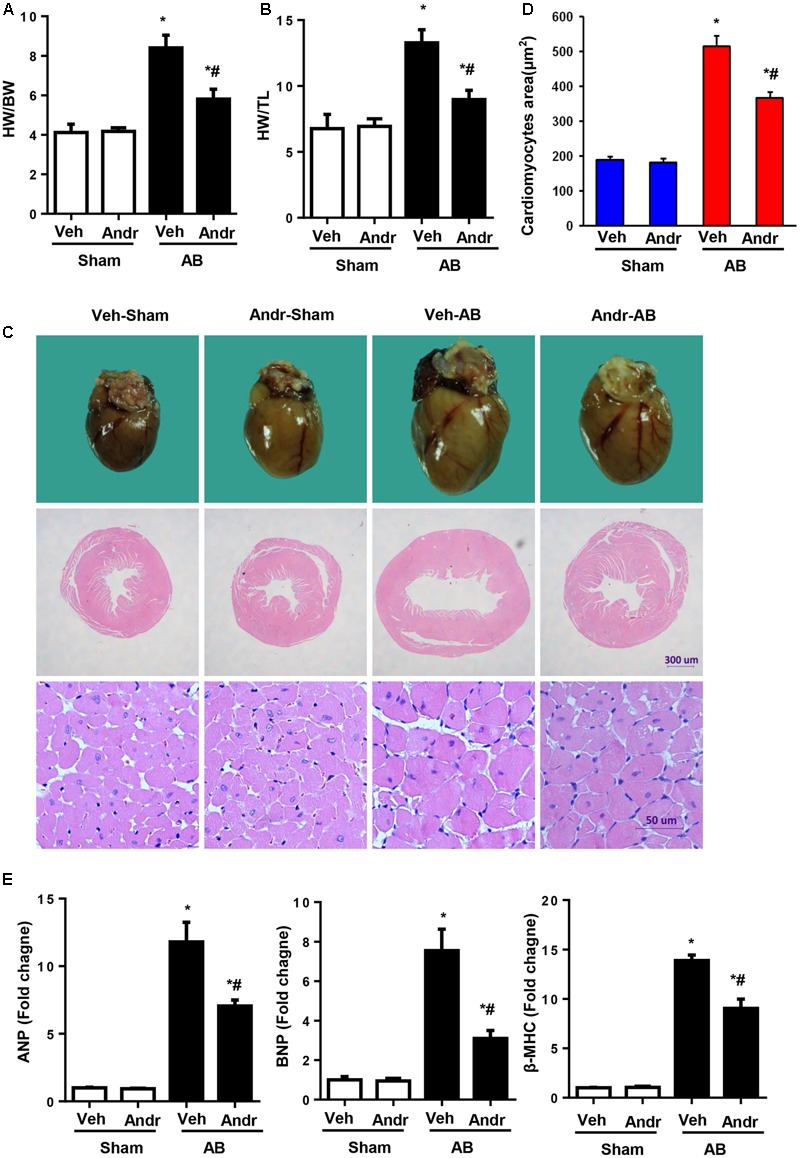
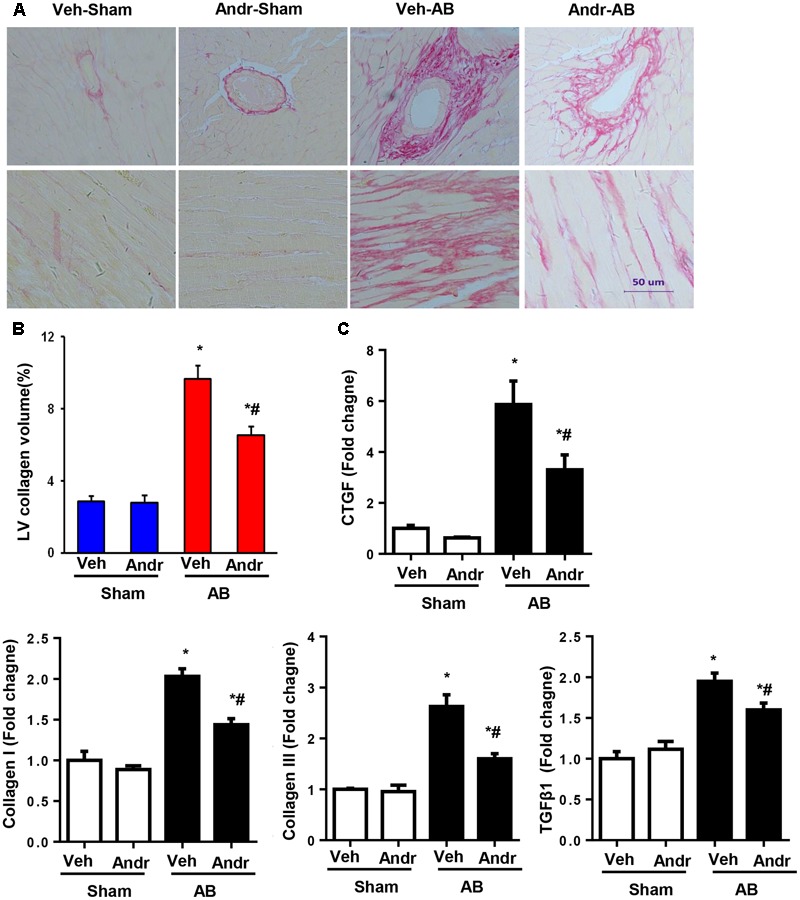
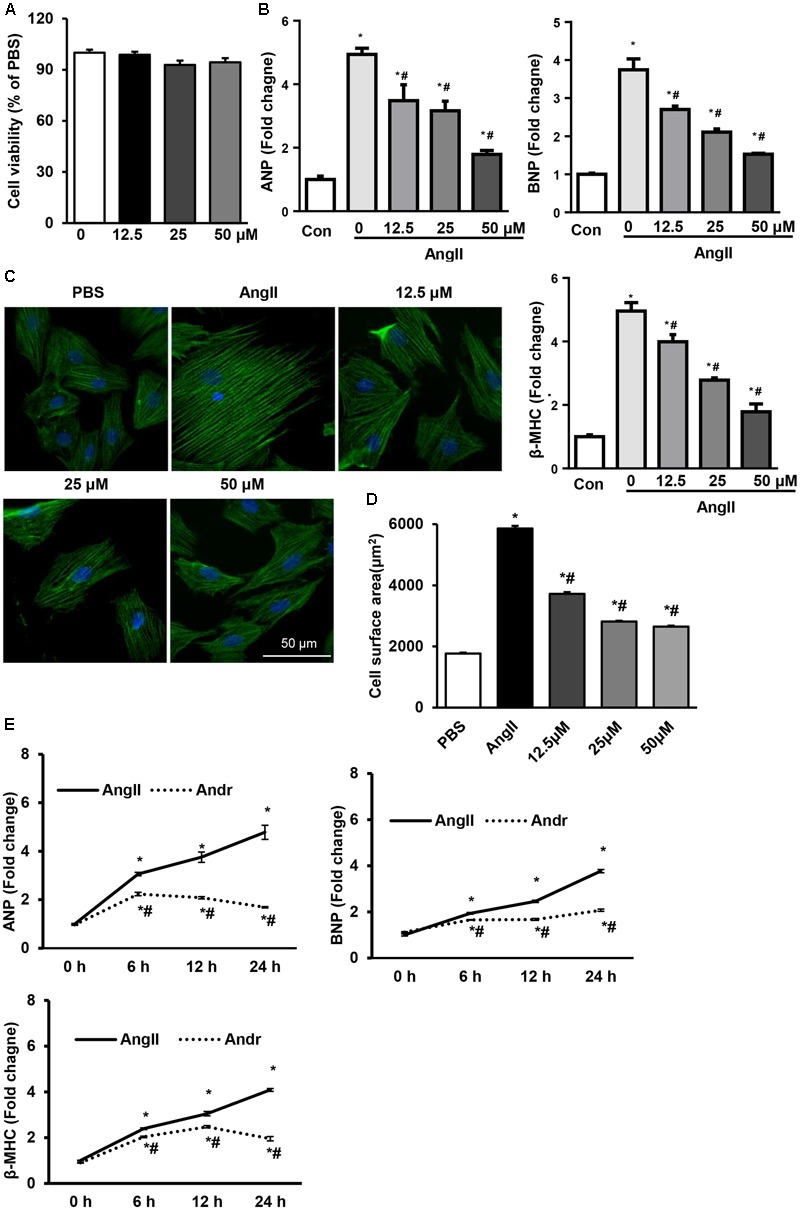
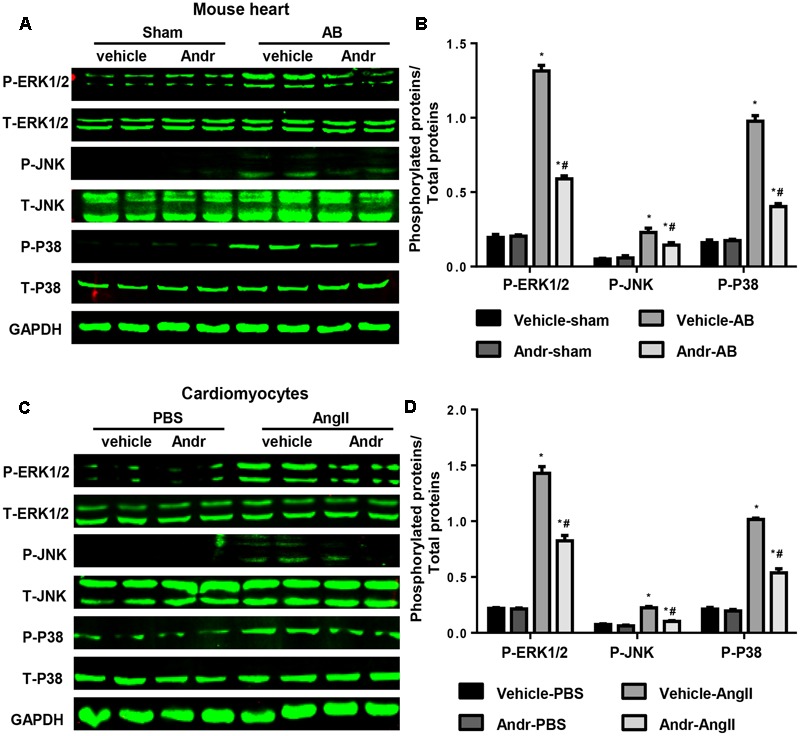
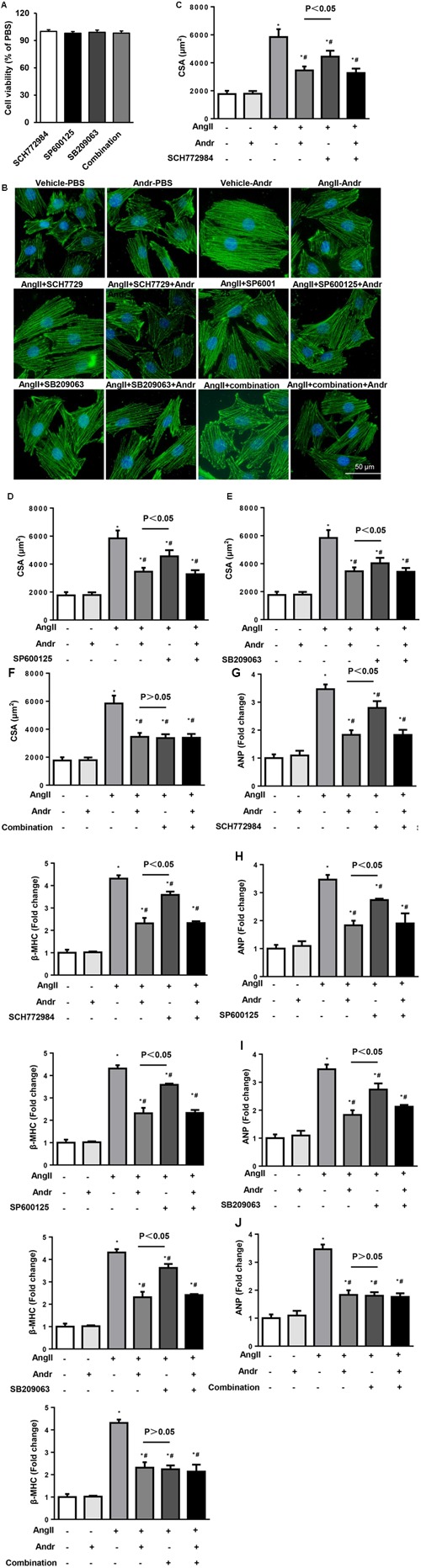
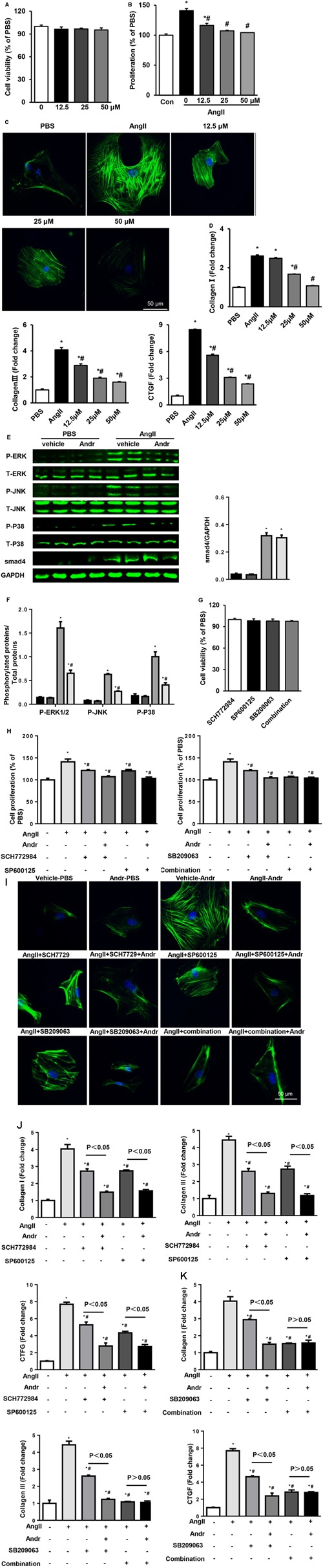
Despite therapeutic advances, heart failure-related mortality rates remain high. Therefore, understanding the pathophysiological mechanisms involved in the remodeling process is crucial for the development of new therapeutic strategies. Andrographolide (Andr), a botanical compound, has potent cardio-protective effects due to its ability to inhibit mitogen-activated protein kinases (MAPKs). Andr has also been shown to inhibit inflammation and apoptosis, which are factors related to cardiac hypertrophy. Our aim was to evaluate the effects of Andr on cardiac hypertrophy and MAPKs activation. Thus, mice were subjected to aortic banding (AB) with/without Andr administration (25 mg/kg/day, orally). Cardiac function was accessed by echocardiography and hemodynamic parameters. Our results showed that Andr administration for 7 weeks decreased cardiac dysfunction and attenuated cardiac hypertrophy and fibrosis in AB mice. Andr treatment induced a strong reduction in the transcription of both hypertrophy (ANP, BNP, and β-MHC) and fibrosis related genes (collagen I, collagen III, CTGF, and TGFβ). In addition, cardiomyocytes treated with Andr showed a reduced hypertrophic response to angiotensin II. Andr significantly inhibited MAPKs activation in both mouse hearts and cardiomyocytes. Treatment with a combination of MAPKs activators abolished the protective effects of Andr in cardiomyocytes. Furthermore, we found that Andr also inhibited the activation of cardiac fibroblasts via the MAPKs pathway, which was confirmed by the application of MAPKs inhibitors. In conclusion, Andr was found to confer a protective effect against experimental cardiac hypertrophy in mice, suggesting its potential as a novel therapeutic drug for pathological cardiac hypertrophy.
Keywords: MAPKs; andrographolide; cardiac hypertrophy; cardiomyocyte; fibroblast.
Figures
Similar articles
-
Andrographolide contributes to the attenuation of cardiac hypertrophy by suppressing endoplasmic reticulum stress.Pharm Biol. 2023 Dec;61(1):61-68. doi: 10.1080/13880209.2022.2157021. Pharm Biol. 2023. PMID: 36548192 Free PMC article.
-
Loss of LRRC25 accelerates pathological cardiac hypertrophy through promoting fibrosis and inflammation regulated by TGF-β1.Biochem Biophys Res Commun. 2018 Nov 17;506(1):137-144. doi: 10.1016/j.bbrc.2018.09.065. Epub 2018 Oct 16. Biochem Biophys Res Commun. 2018. PMID: 30340835
-
BRD4 blockage alleviates pathological cardiac hypertrophy through the suppression of fibrosis and inflammation via reducing ROS generation.Biomed Pharmacother. 2020 Jan;121:109368. doi: 10.1016/j.biopha.2019.109368. Epub 2019 Nov 25. Biomed Pharmacother. 2020. PMID: 31707348
-
RIP2 deficiency attenuates cardiac hypertrophy, inflammation and fibrosis in pressure overload induced mice.Biochem Biophys Res Commun. 2017 Nov 18;493(2):1151-1158. doi: 10.1016/j.bbrc.2017.07.035. Epub 2017 Jul 8. Biochem Biophys Res Commun. 2017. PMID: 28698147
-
Regulator of G-Protein Signaling 10 Negatively Regulates Cardiac Remodeling by Blocking Mitogen-Activated Protein Kinase-Extracellular Signal-Regulated Protein Kinase 1/2 Signaling.Hypertension. 2016 Jan;67(1):86-98. doi: 10.1161/HYPERTENSIONAHA.115.05957. Epub 2015 Nov 16. Hypertension. 2016. PMID: 26573707 Review.
Cited by
-
Cordycepin ameliorates cardiac hypertrophy via activating the AMPKα pathway.J Cell Mol Med. 2019 Aug;23(8):5715-5727. doi: 10.1111/jcmm.14485. Epub 2019 Jun 21. J Cell Mol Med. 2019. PMID: 31225721 Free PMC article.
-
Promising Therapeutic Treatments for Cardiac Fibrosis: Herbal Plants and Their Extracts.Cardiol Ther. 2023 Sep;12(3):415-443. doi: 10.1007/s40119-023-00319-4. Epub 2023 May 29. Cardiol Ther. 2023. PMID: 37247171 Free PMC article. Review.
-
Andrographolide Protects against HG-Induced Inflammation, Apoptosis, Migration, and Impairment of Angiogenesis via PI3K/AKT-eNOS Signalling in HUVECs.Mediators Inflamm. 2019 Oct 7;2019:6168340. doi: 10.1155/2019/6168340. eCollection 2019. Mediators Inflamm. 2019. PMID: 31686985 Free PMC article.
-
Andrographolide protects against atrial fibrillation by alleviating oxidative stress injury and promoting impaired mitochondrial bioenergetics.J Zhejiang Univ Sci B. 2023 Jul 15;24(7):632-649. doi: 10.1631/jzus.B2300086. J Zhejiang Univ Sci B. 2023. PMID: 37455139 Free PMC article.
-
Andrographolide Protects Against Adverse Cardiac Remodeling After Myocardial Infarction through Enhancing Nrf2 Signaling Pathway.Int J Biol Sci. 2020 Jan 1;16(1):12-26. doi: 10.7150/ijbs.37269. eCollection 2020. Int J Biol Sci. 2020. PMID: 31892842 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous