

Genetic diversity of the African malaria vector Anopheles gambiae
- PMID: 29186111
- PMCID: PMC6026373
- DOI: 10.1038/nature24995
Genetic diversity of the African malaria vector Anopheles gambiae
Abstract
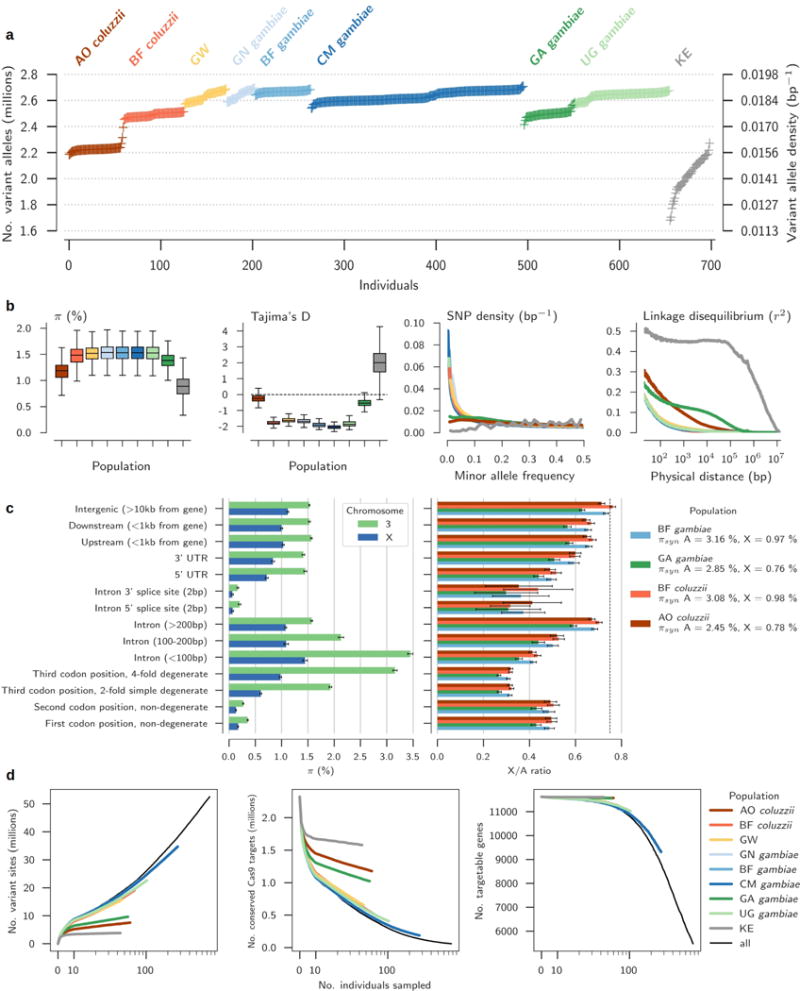
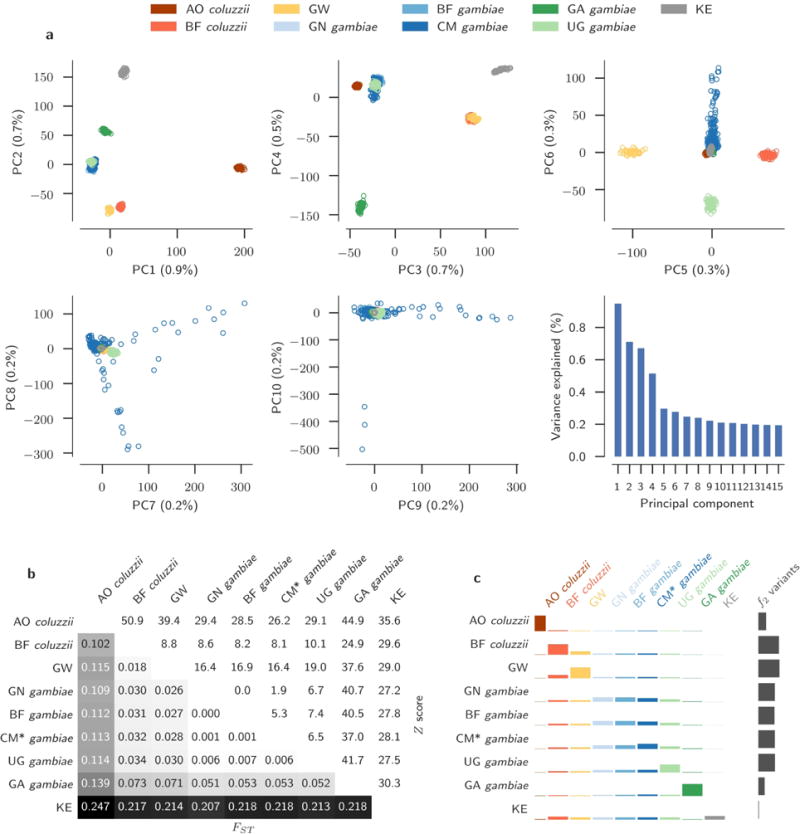
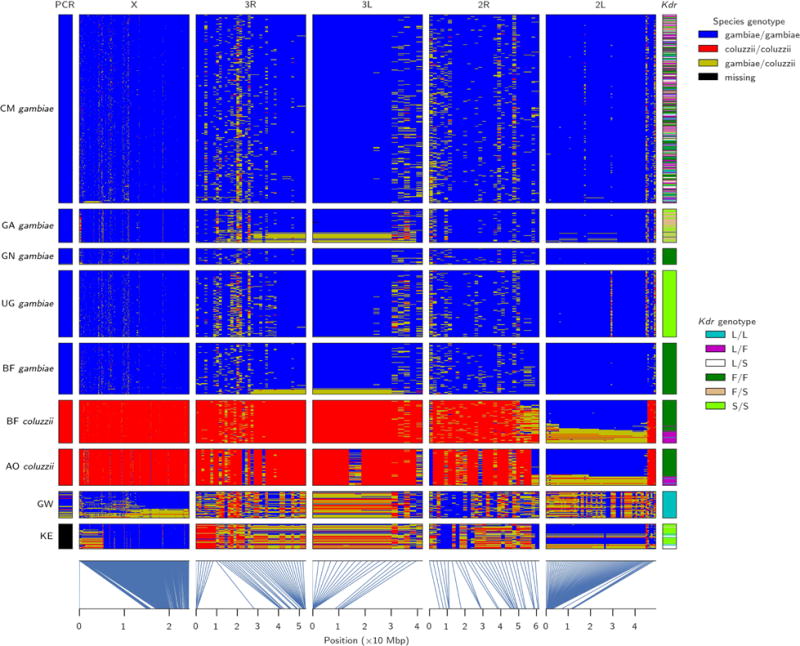
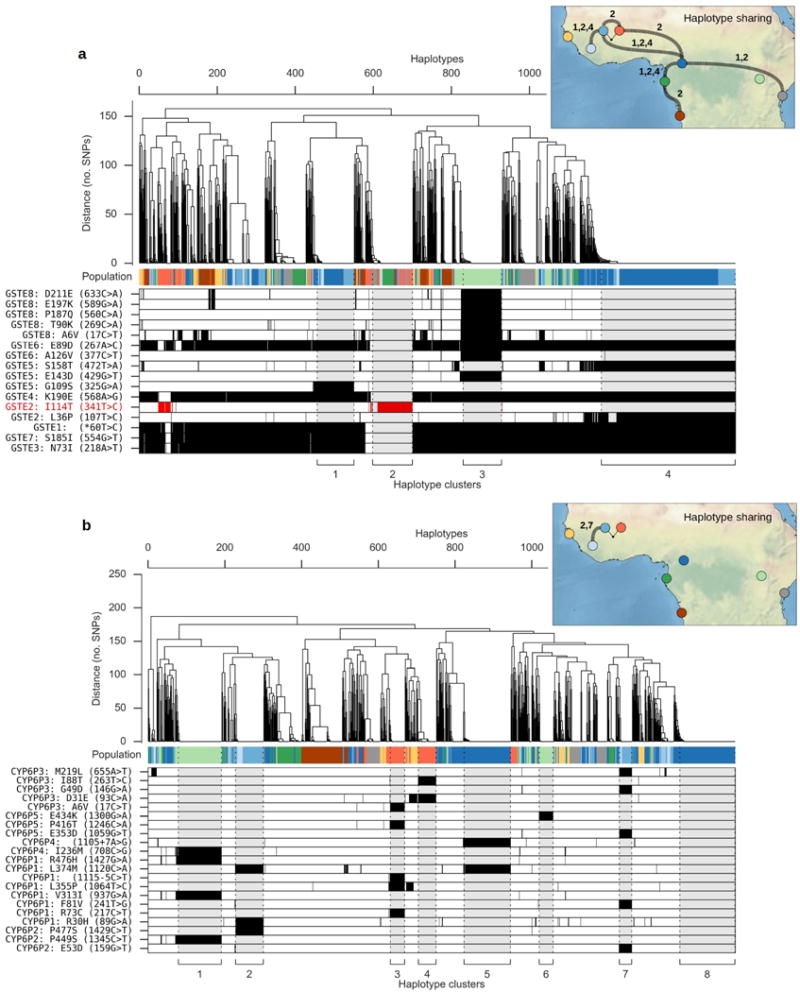
The sustainability of malaria control in Africa is threatened by the rise of insecticide resistance in Anopheles mosquitoes, which transmit the disease. To gain a deeper understanding of how mosquito populations are evolving, here we sequenced the genomes of 765 specimens of Anopheles gambiae and Anopheles coluzzii sampled from 15 locations across Africa, and identified over 50 million single nucleotide polymorphisms within the accessible genome. These data revealed complex population structure and patterns of gene flow, with evidence of ancient expansions, recent bottlenecks, and local variation in effective population size. Strong signals of recent selection were observed in insecticide-resistance genes, with several sweeps spreading over large geographical distances and between species. The design of new tools for mosquito control using gene-drive systems will need to take account of high levels of genetic diversity in natural mosquito populations.
Conflict of interest statement
The authors declare no competing financial interests.
Figures









References
-
- Torre A della, et al. Molecular evidence of incipient speciation within Anopheles gambiae s.s. in West Africa. Insect Mol Biol. 2001;10:9–18. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
