

Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control
- PMID: 29186125
- PMCID: PMC6217924
- DOI: 10.1038/nature24678
Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control
Abstract
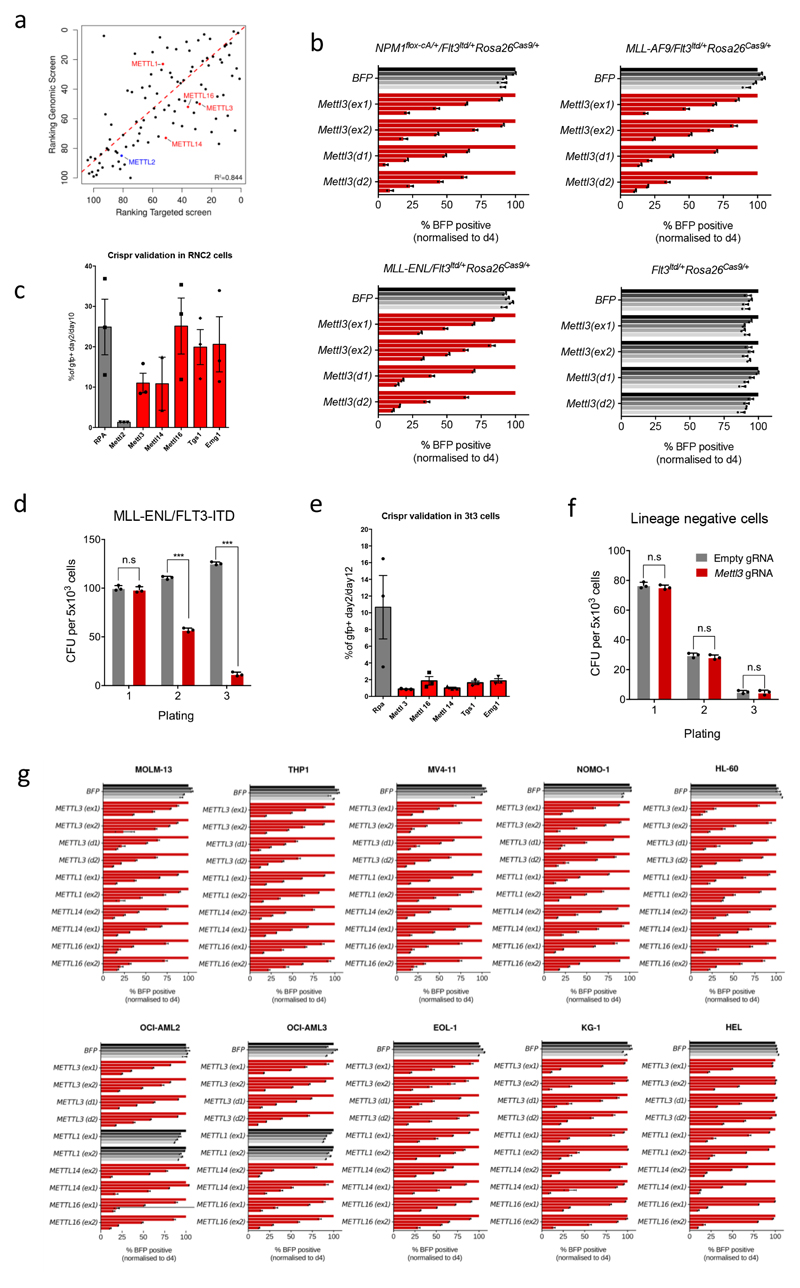
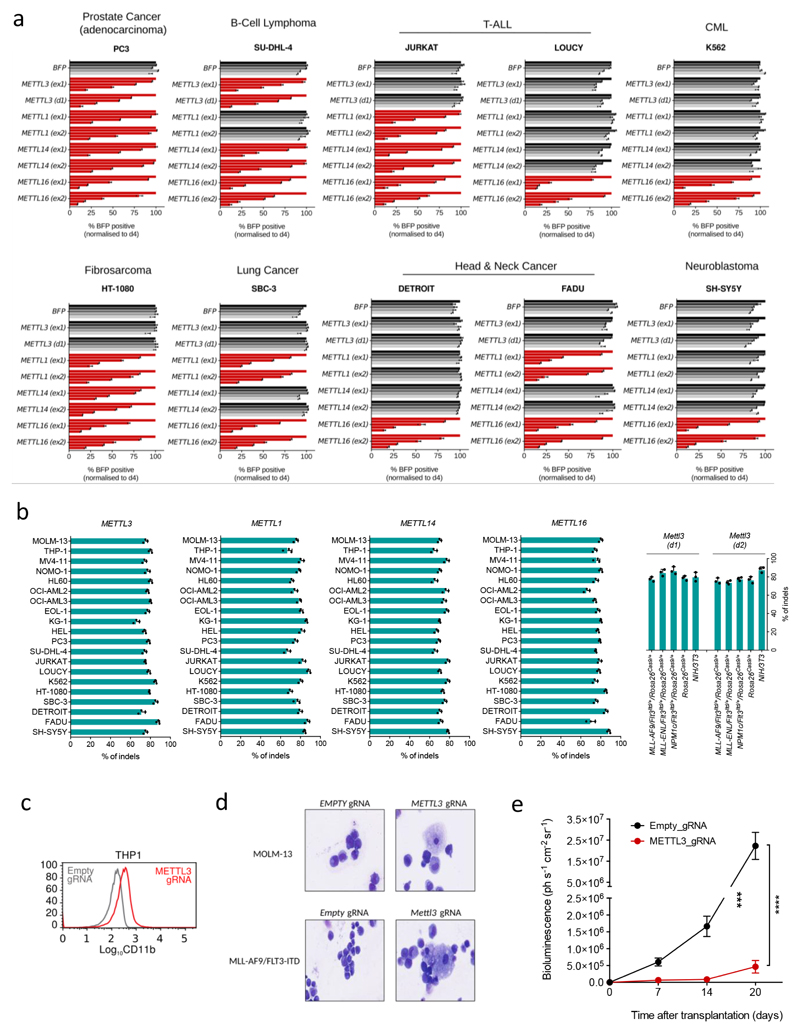
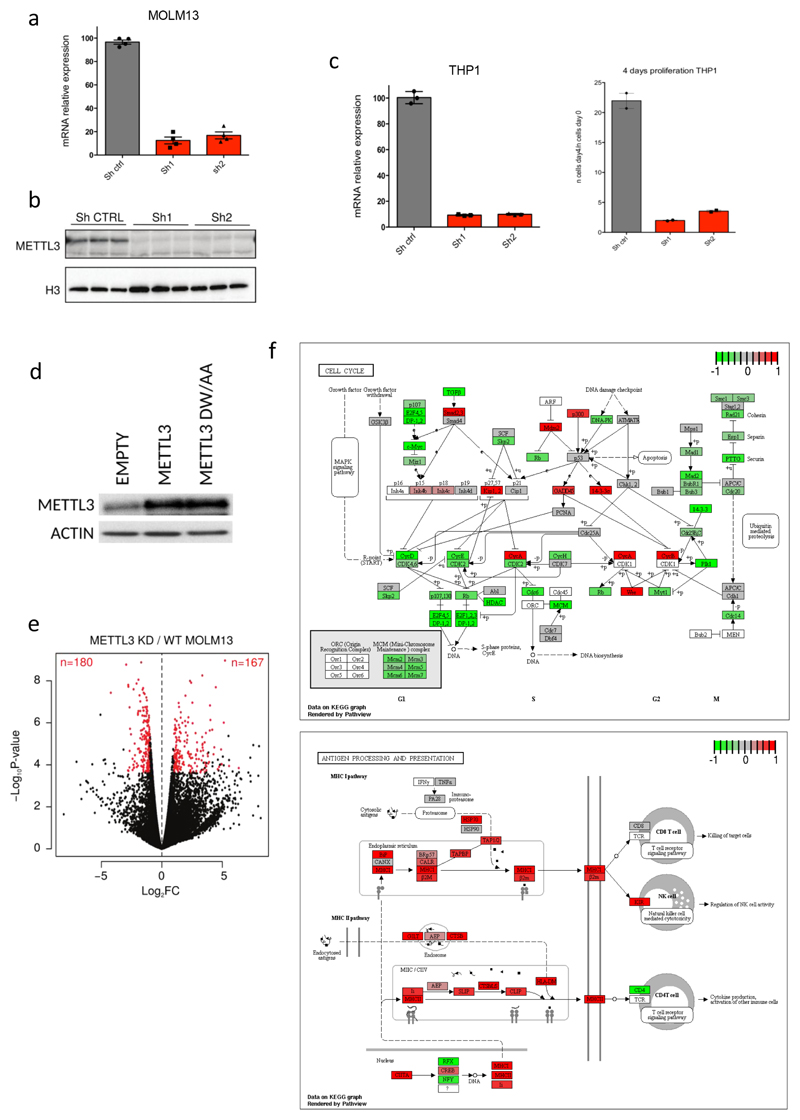
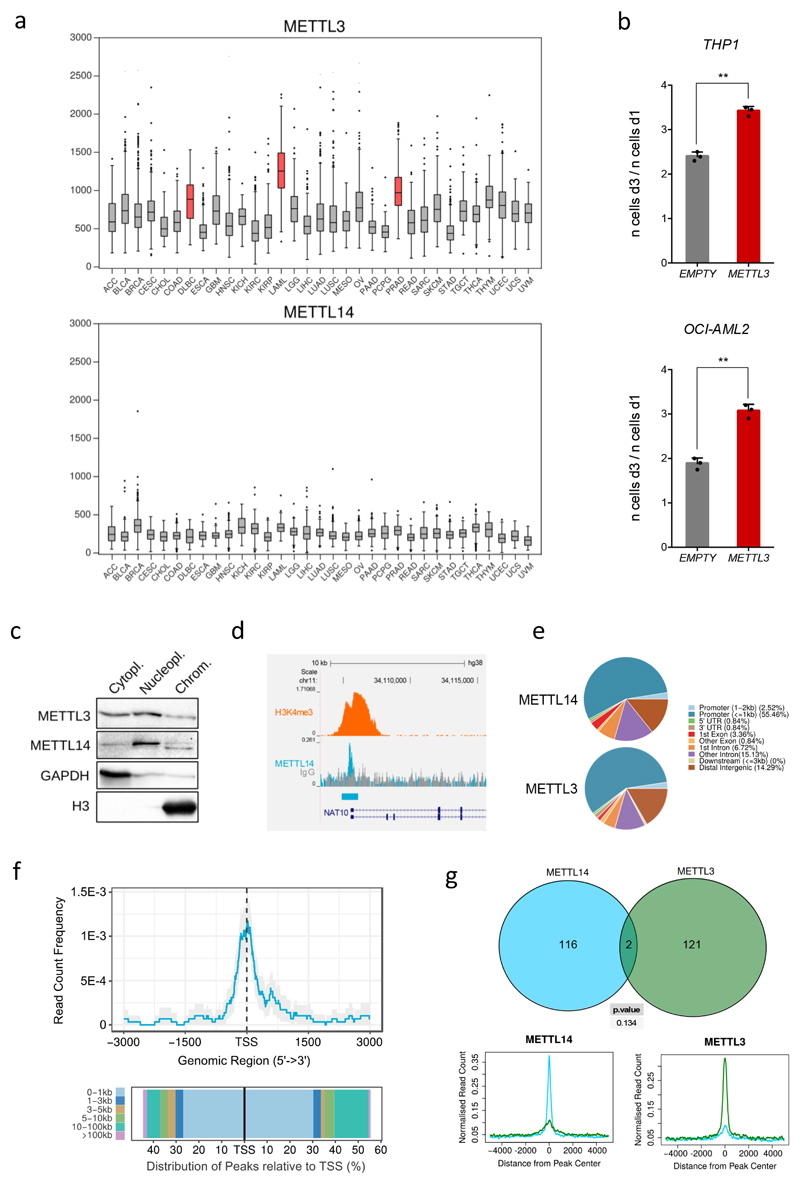
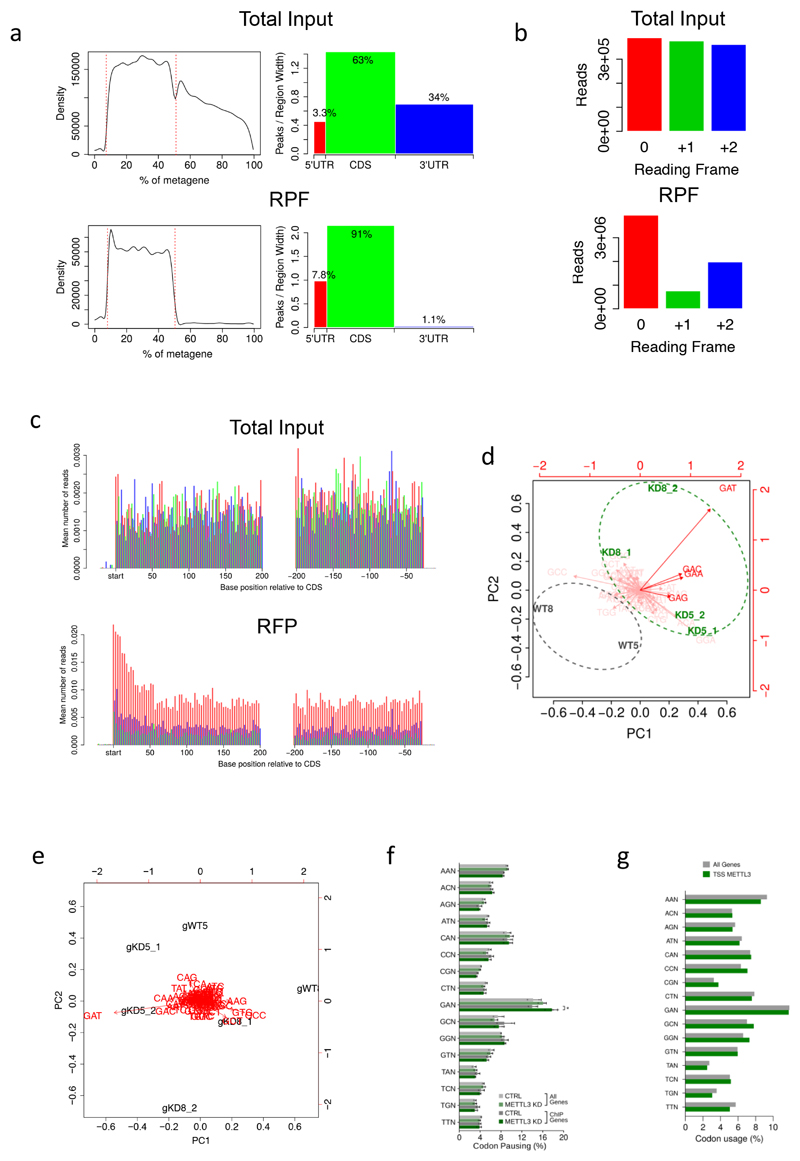
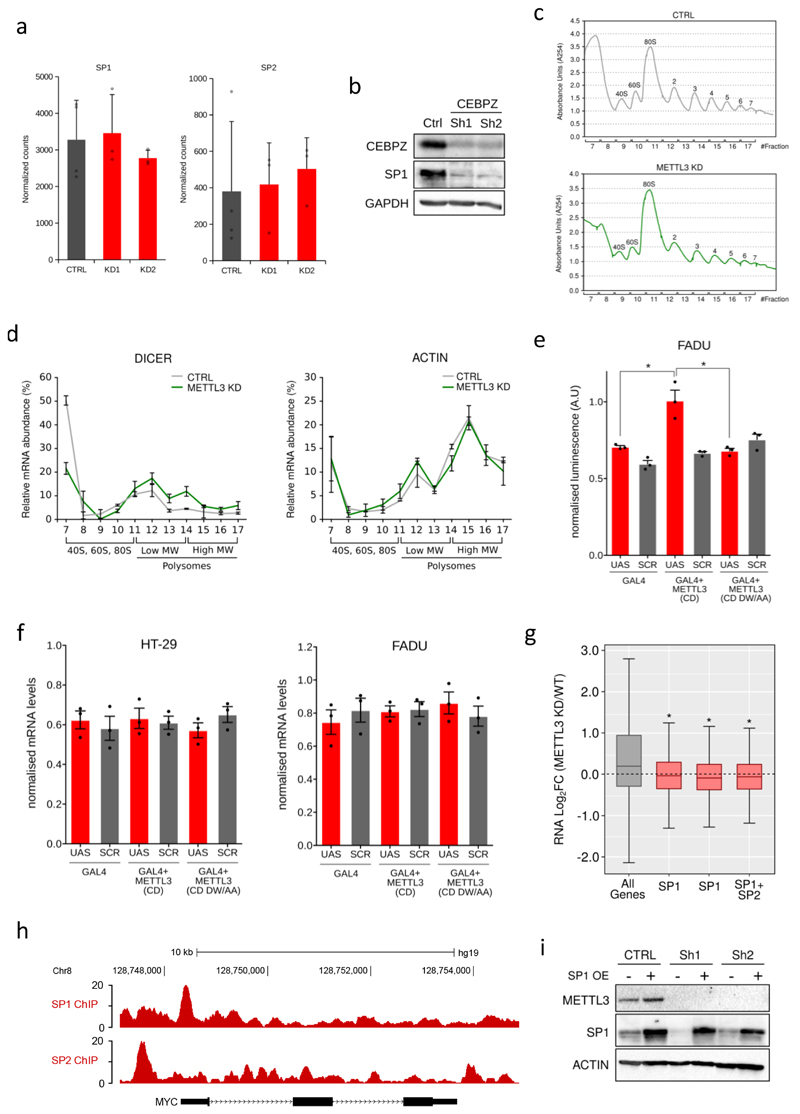
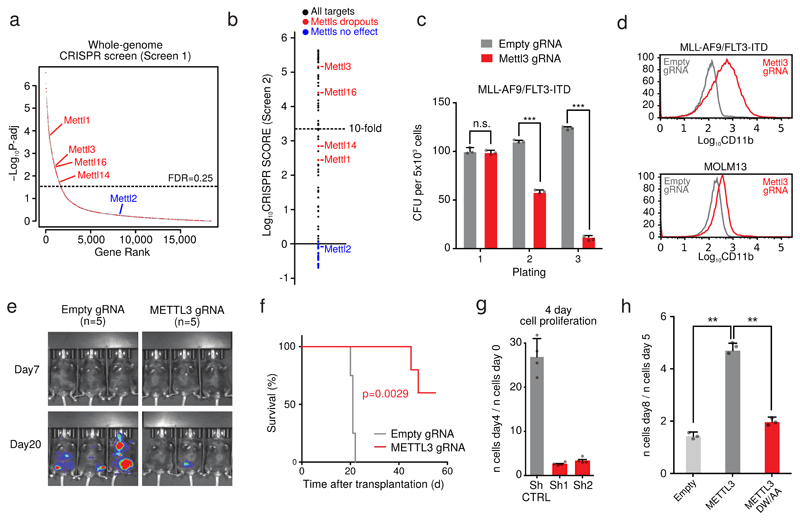
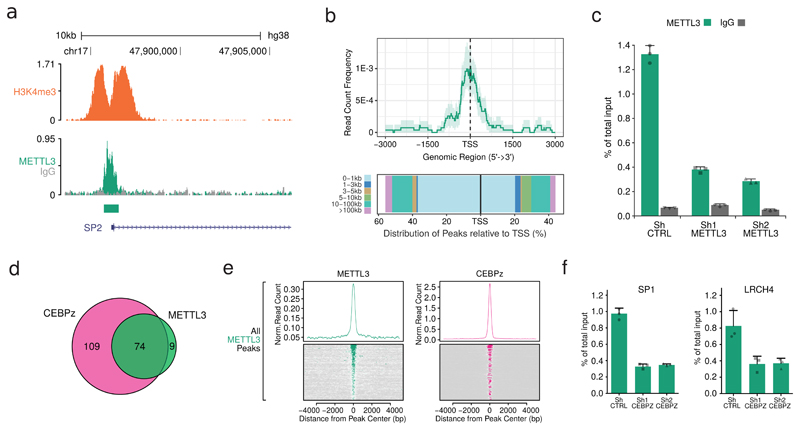
N6-methyladenosine (m6A) is an abundant internal RNA modification in both coding and non-coding RNAs that is catalysed by the METTL3-METTL14 methyltransferase complex. However, the specific role of these enzymes in cancer is still largely unknown. Here we define a pathway that is specific for METTL3 and is implicated in the maintenance of a leukaemic state. We identify METTL3 as an essential gene for growth of acute myeloid leukaemia cells in two distinct genetic screens. Downregulation of METTL3 results in cell cycle arrest, differentiation of leukaemic cells and failure to establish leukaemia in immunodeficient mice. We show that METTL3, independently of METTL14, associates with chromatin and localizes to the transcriptional start sites of active genes. The vast majority of these genes have the CAATT-box binding protein CEBPZ present at the transcriptional start site, and this is required for recruitment of METTL3 to chromatin. Promoter-bound METTL3 induces m6A modification within the coding region of the associated mRNA transcript, and enhances its translation by relieving ribosome stalling. We show that genes regulated by METTL3 in this way are necessary for acute myeloid leukaemia. Together, these data define METTL3 as a regulator of a chromatin-based pathway that is necessary for maintenance of the leukaemic state and identify this enzyme as a potential therapeutic target for acute myeloid leukaemia.
Conflict of interest statement
T.K. is a co-founder of Abcam Plc and Storm Therapeutics Ltd, Cambridge, UK.
A.H. is an employee of Storm Therapeutics Ltd, Cambridge, UK.
Figures
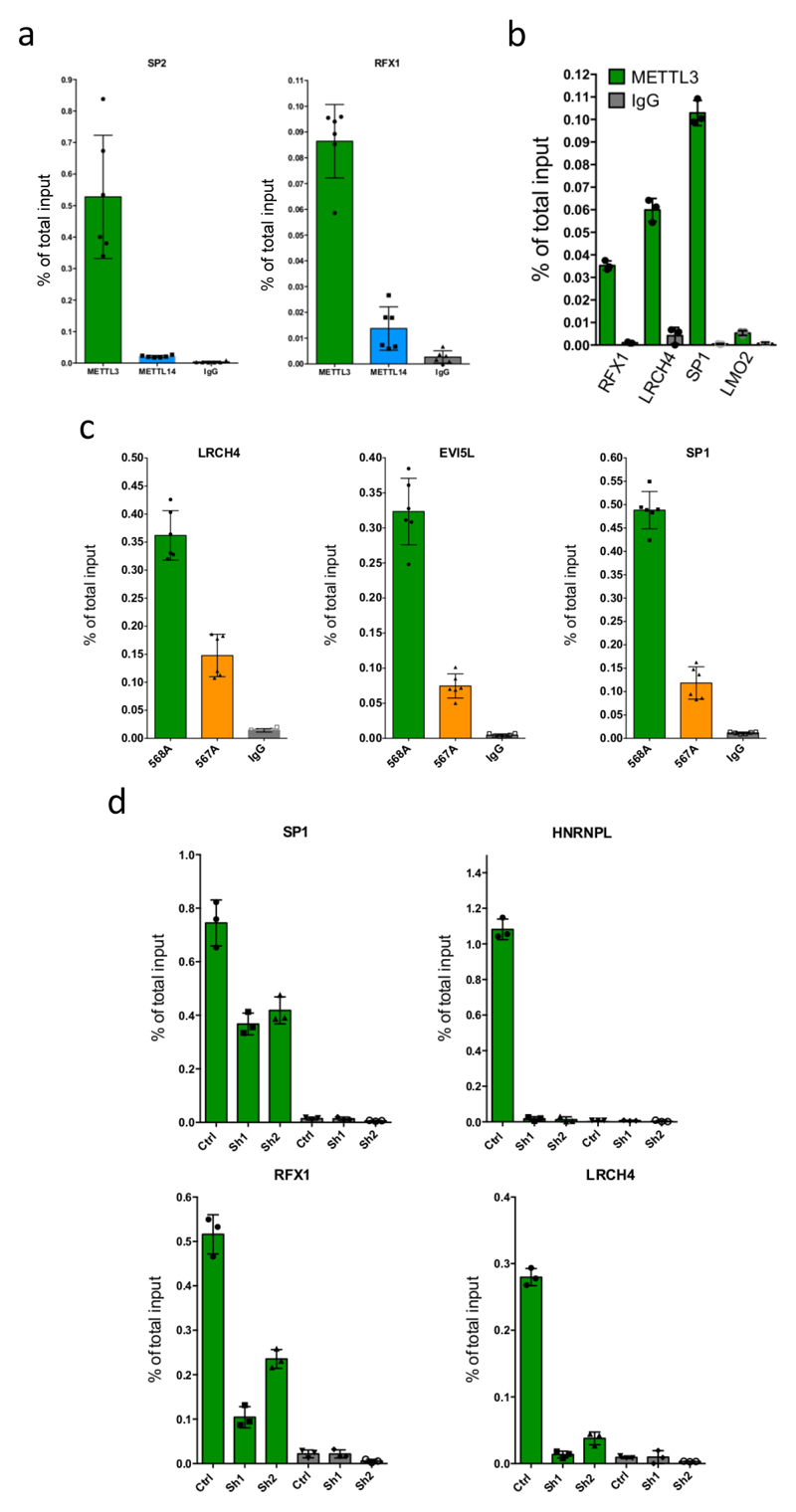
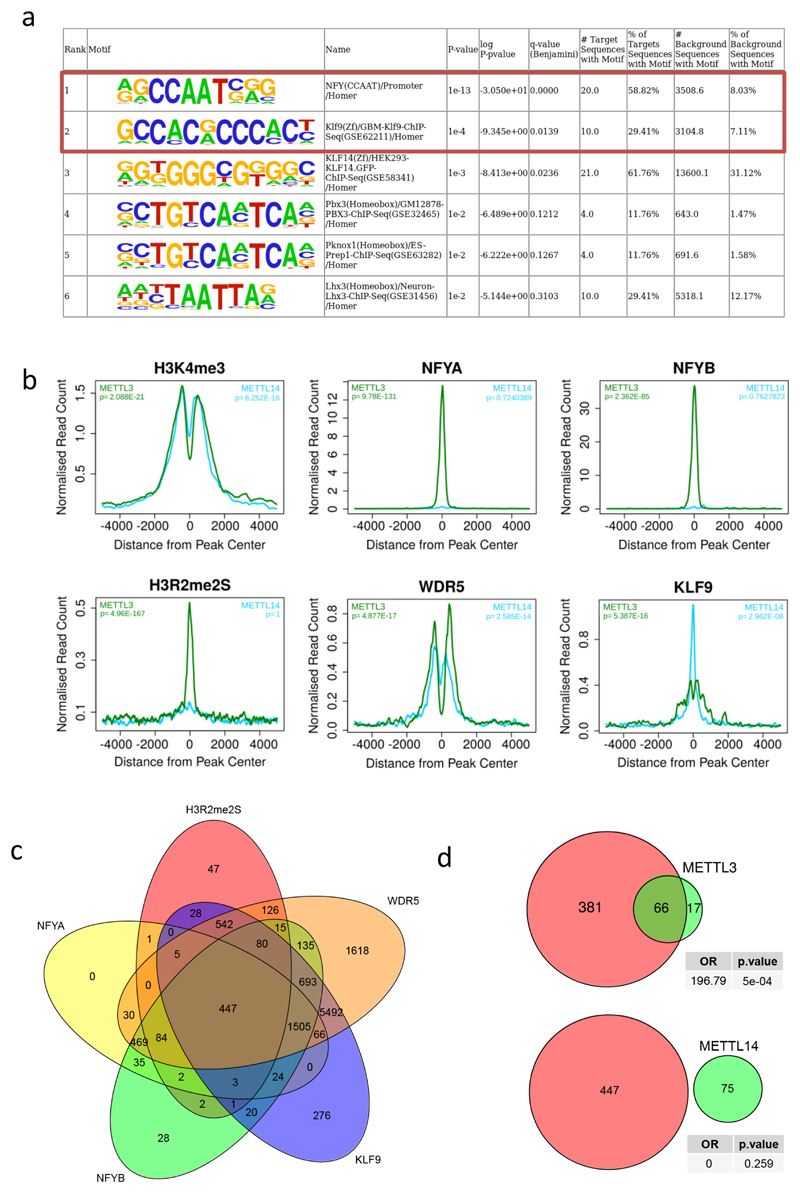
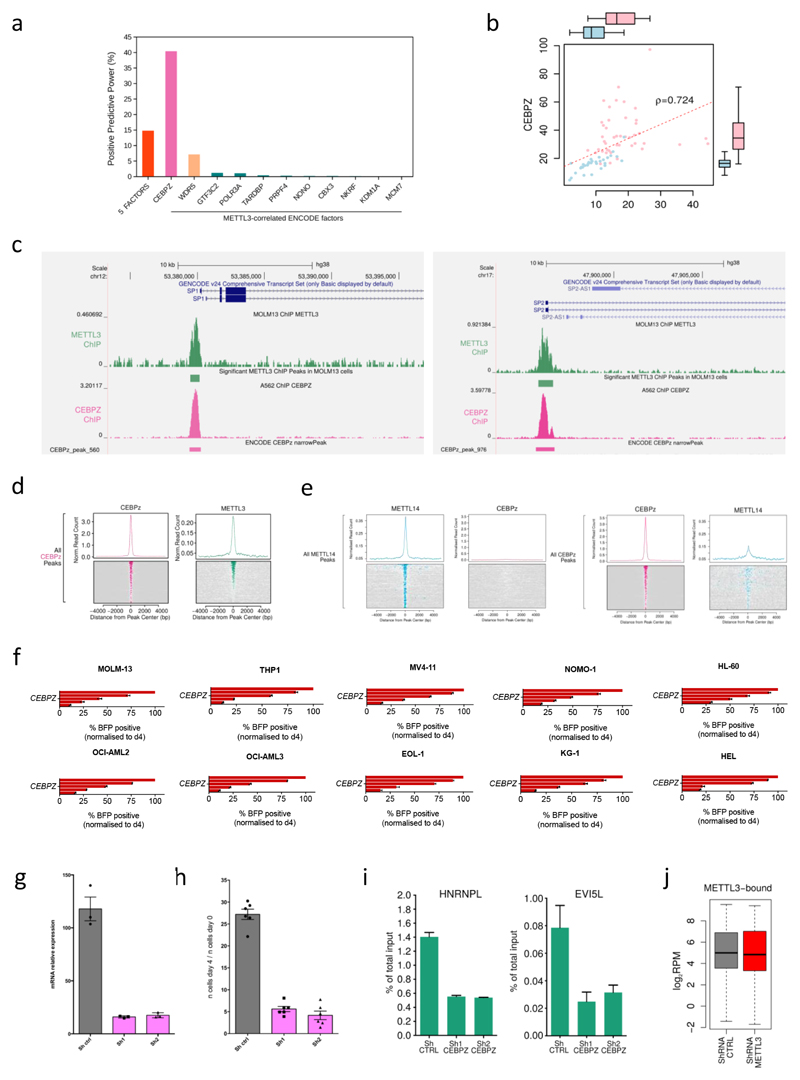
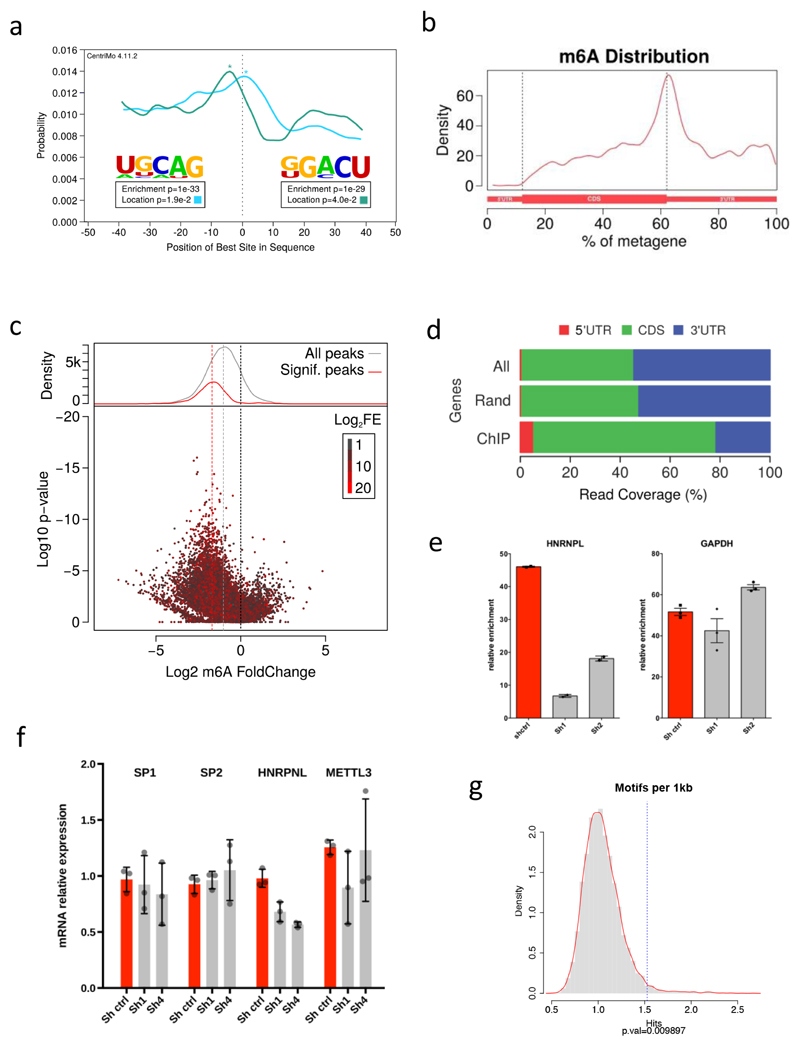
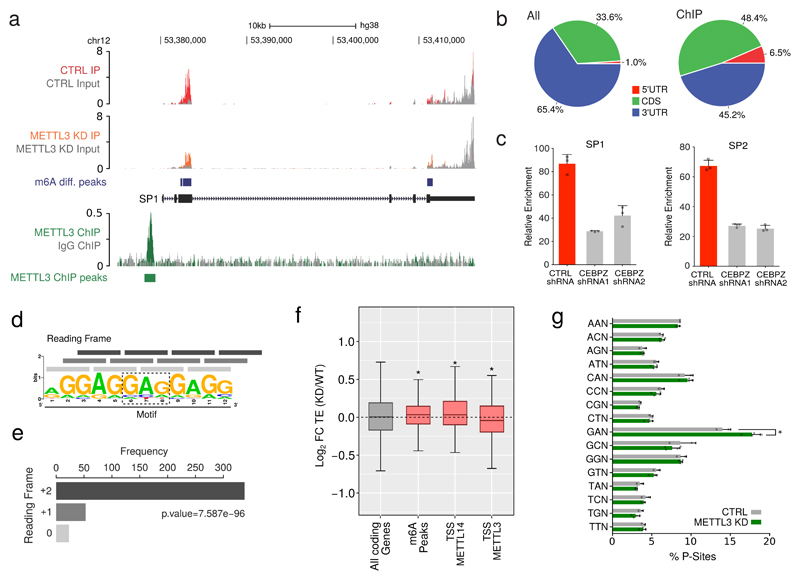
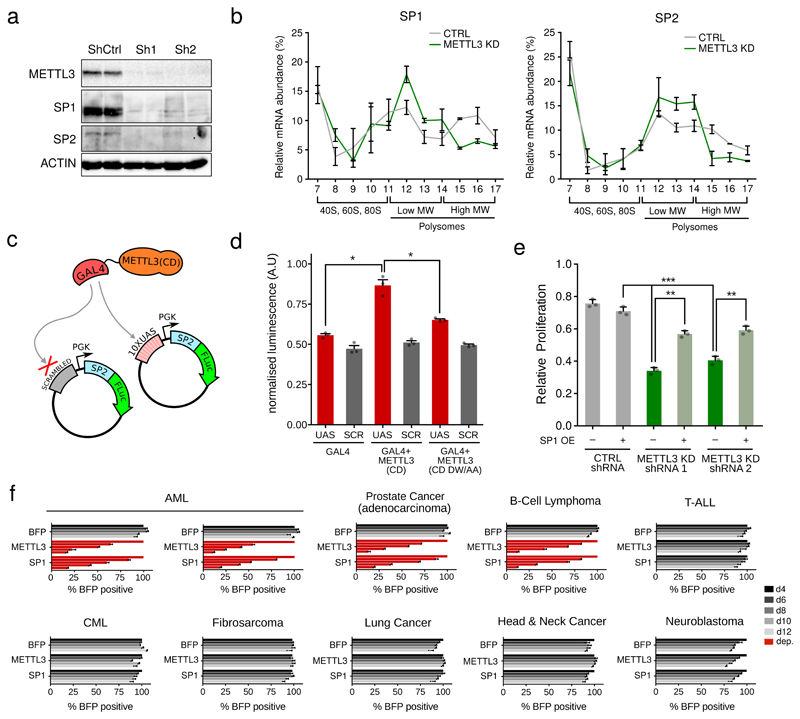
Comment in
-
Meddling with METTLs in Normal and Leukemia Stem Cells.Cell Stem Cell. 2018 Feb 1;22(2):139-141. doi: 10.1016/j.stem.2018.01.013. Cell Stem Cell. 2018. PMID: 29395048
References
-
- Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- WT095663MA/WT_/Wellcome Trust/United Kingdom
- 092096/WT_/Wellcome Trust/United Kingdom
- 10827/CRUK_/Cancer Research UK/United Kingdom
- 098051/WT_/Wellcome Trust/United Kingdom
- 095663/WT_/Wellcome Trust/United Kingdom
- 23015/CRUK_/Cancer Research UK/United Kingdom
- A17001/CRUK_/Cancer Research UK/United Kingdom
- 17001/CRUK_/Cancer Research UK/United Kingdom
- 268569/ERC_/European Research Council/International
- A23015/CRUK_/Cancer Research UK/United Kingdom
- MC_PC_12009/MRC_/Medical Research Council/United Kingdom
- C6946/AI4492/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
