

PHYLOSCANNER: Inferring Transmission from Within- and Between-Host Pathogen Genetic Diversity
- PMID: 29186559
- PMCID: PMC5850600
- DOI: 10.1093/molbev/msx304
PHYLOSCANNER: Inferring Transmission from Within- and Between-Host Pathogen Genetic Diversity
Abstract
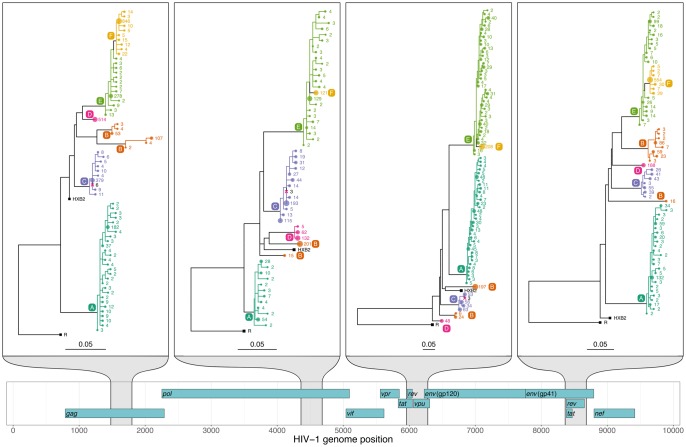
A central feature of pathogen genomics is that different infectious particles (virions and bacterial cells) within an infected individual may be genetically distinct, with patterns of relatedness among infectious particles being the result of both within-host evolution and transmission from one host to the next. Here, we present a new software tool, phyloscanner, which analyses pathogen diversity from multiple infected hosts. phyloscanner provides unprecedented resolution into the transmission process, allowing inference of the direction of transmission from sequence data alone. Multiply infected individuals are also identified, as they harbor subpopulations of infectious particles that are not connected by within-host evolution, except where recombinant types emerge. Low-level contamination is flagged and removed. We illustrate phyloscanner on both viral and bacterial pathogens, namely HIV-1 sequenced on Illumina and Roche 454 platforms, HCV sequenced with the Oxford Nanopore MinION platform, and Streptococcus pneumoniae with sequences from multiple colonies per individual. phyloscanner is available from https://github.com/BDI-pathogens/phyloscanner.
Keywords: molecular epidemiology; multiple infection; pathogen diversity; pathogen genomics; pathogen transmission; phylogenetics.
© The Author 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures








References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410. - PubMed
-
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120.http://dx.doi.org/10.1093/bioinformatics/btu170 - DOI - PMC - PubMed
-
- Bonsall D, Ansari MA, Ip C, Trebes A, Brown A, Klenerman P, Buck D, null N, Piazza P, Barnes E, et al.2015. ve-SEQ: robust, unbiased enrichment for streamlined detection and whole-genome sequencing of HCV and other highly diverse pathogens [version 1; referees: 2 approved, 1 approved with reservations]. F1000Research 4: 1062. - PMC - PubMed
-
- Cornelissen M, Gall A, Vink M, Zorgdrager F, Binter Š, Edwards S, Jurriaans S, Bakker M, Ong SH, Gras L, et al.2016. From clinical sample to complete genome: comparing methods for the extraction of HIV-1 RNA for high-throughput deep sequencing. Virus Res. 239:10–16. - PubMed
-
- Cornelissen M, Pasternak AO, Grijsen ML, Zorgdrager F, Bakker M, Blom P, Prins JM, Jurriaans S, van der Kuyl AC.. 2012. HIV-1 dual infection is associated with faster CD4+ T-cell decline in a cohort of men with primary HIV infection. Clin Infect Dis. 54(4):539.. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
