

Relaxed Selection During a Recent Human Expansion
- PMID: 29187508
- PMCID: PMC5788536
- DOI: 10.1534/genetics.117.300551
Relaxed Selection During a Recent Human Expansion
Abstract
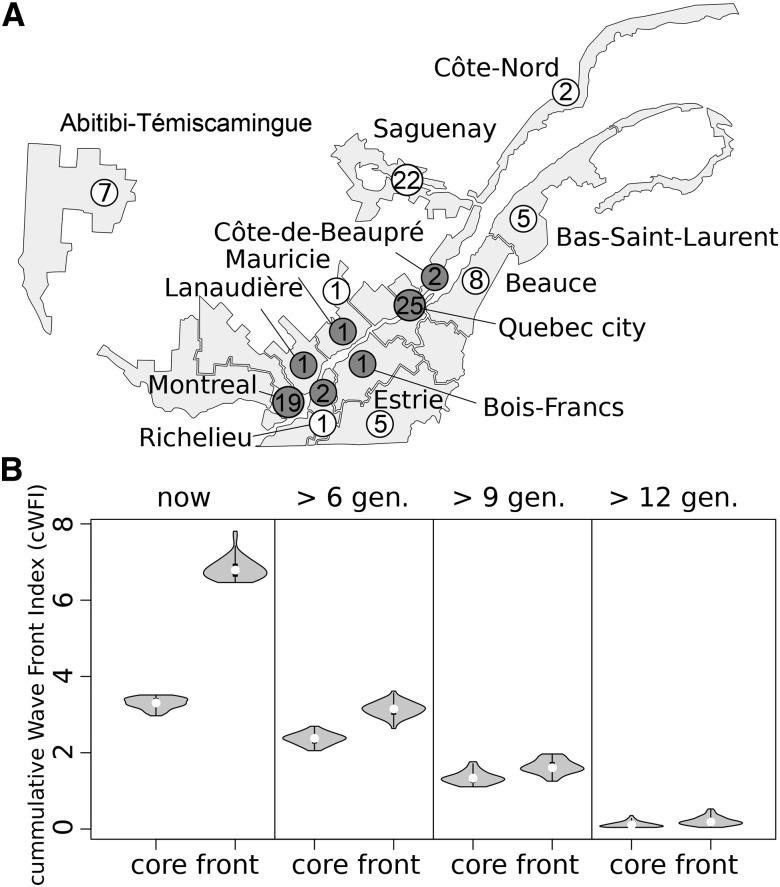
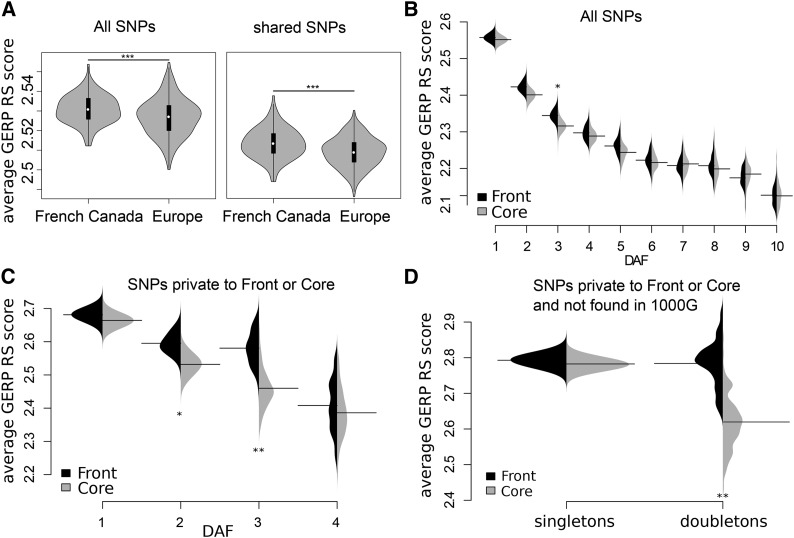
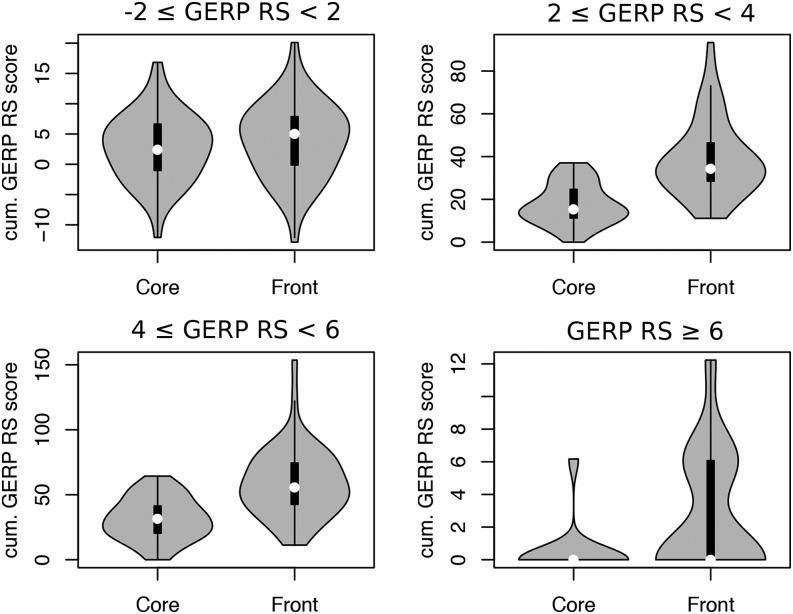
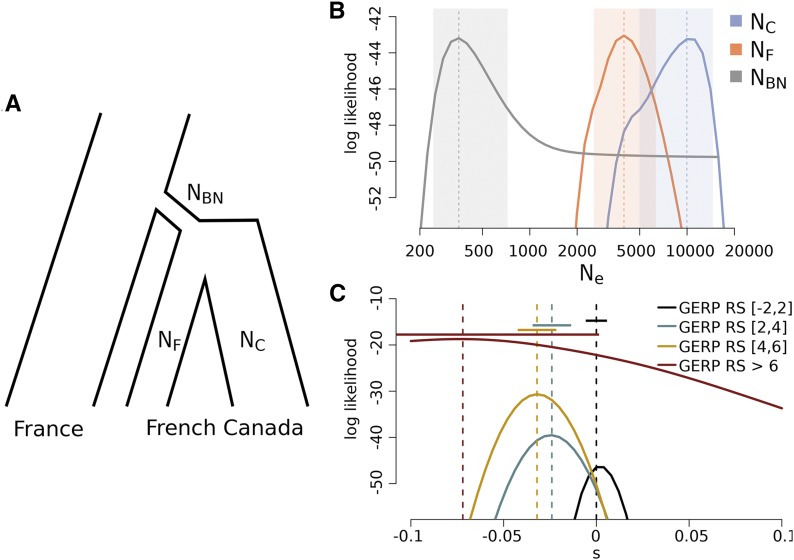
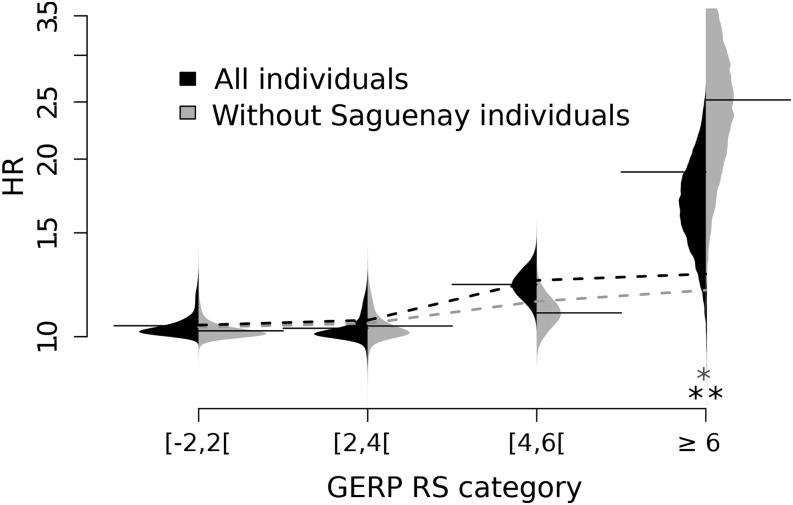
Humans have colonized the planet through a series of range expansions, which deeply impacted genetic diversity in newly settled areas and potentially increased the frequency of deleterious mutations on expanding wave fronts. To test this prediction, we studied the genomic diversity of French Canadians who colonized Quebec in the 17th century. We used historical information and records from ∼4000 ascending genealogies to select individuals whose ancestors lived mostly on the colonizing wave front and individuals whose ancestors remained in the core of the settlement. Comparison of exomic diversity reveals that: (i) both new and low-frequency variants are significantly more deleterious in front than in core individuals, (ii) equally deleterious mutations are at higher frequencies in front individuals, and (iii) front individuals are two times more likely to be homozygous for rare very deleterious mutations present in Europeans. These differences have emerged in the past six to nine generations and cannot be explained by differential inbreeding, but are consistent with relaxed selection mainly due to higher rates of genetic drift on the wave front. Demographic inference and modeling of the evolution of rare variants suggest lower effective size on the front, and lead to an estimation of selection coefficients that increase with conservation scores. Even though range expansions have had a relatively limited impact on the overall fitness of French Canadians, they could explain the higher prevalence of recessive genetic diseases in recently settled regions of Quebec.
Keywords: Quebec; genetic drift; mutation load; range expansion.
Copyright © 2018 by the Genetics Society of America.
Figures
References
-
- Alexa A., Rahnenfuhrer J., Lengauer T., 2006. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22: 1600–1607. - PubMed
-
- Awadalla P., Boileau C., Payette Y., Idaghdour Y., Goulet J. P., et al. , 2013. Cohort profile of the CARTaGENE study: Quebec’s population-based biobank for public health and personalized genomics. Int. J. Epidemiol. 42: 1285–1299. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
