

Microbiome Datasets Are Compositional: And This Is Not Optional
- PMID: 29187837
- PMCID: PMC5695134
- DOI: 10.3389/fmicb.2017.02224
Microbiome Datasets Are Compositional: And This Is Not Optional
Abstract
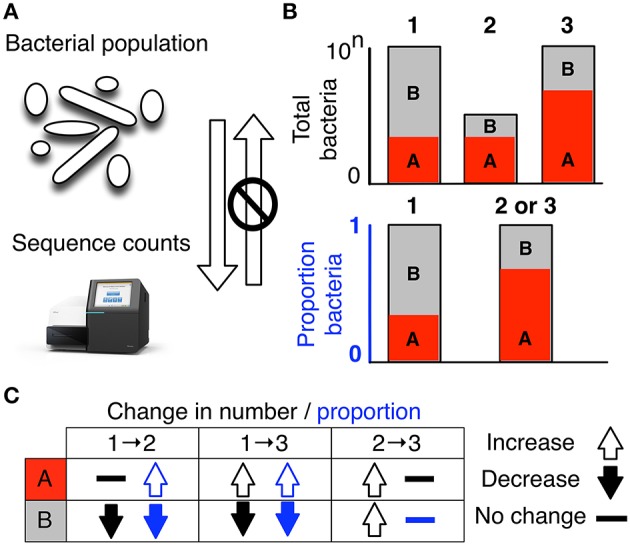
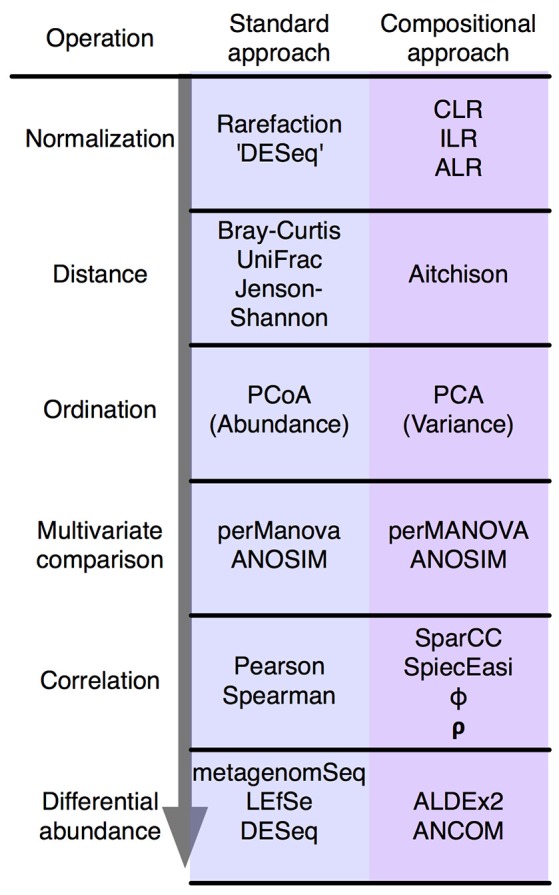
Datasets collected by high-throughput sequencing (HTS) of 16S rRNA gene amplimers, metagenomes or metatranscriptomes are commonplace and being used to study human disease states, ecological differences between sites, and the built environment. There is increasing awareness that microbiome datasets generated by HTS are compositional because they have an arbitrary total imposed by the instrument. However, many investigators are either unaware of this or assume specific properties of the compositional data. The purpose of this review is to alert investigators to the dangers inherent in ignoring the compositional nature of the data, and point out that HTS datasets derived from microbiome studies can and should be treated as compositions at all stages of analysis. We briefly introduce compositional data, illustrate the pathologies that occur when compositional data are analyzed inappropriately, and finally give guidance and point to resources and examples for the analysis of microbiome datasets using compositional data analysis.
Keywords: Bayesian estimation; compositional data; correlation; count normalization; high-throughput sequencing; microbiota; relative abundance.
Figures
References
-
- Aitchison J. (1983). Principal component analysis of compositional data. Biometrika 70, 57–65. 10.1093/biomet/70.1.57 - DOI
-
- Aitchison J. (1986). The Statistical Analysis of Compositional Data. London: Chapman and Hall.
-
- Aitchison J., Barceló-Vidal C., Martín-Fernández J. A., Pawlowsky-Glahn V. (2000). Logratio analysis and compositional distance. Math. Geol. 32, 271–275. 10.1023/A:1007529726302 - DOI
-
- Aitchison J., Greenacre M. (2002). Biplots of compositional data. J. Roy. Stat. Soc. Ser. C 51, 375–392. 10.1111/1467-9876.00275 - DOI
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical