

Quantification of Inter-Sample Differences in T-Cell Receptor Repertoires Using Sequence-Based Information
- PMID: 29187849
- PMCID: PMC5694755
- DOI: 10.3389/fimmu.2017.01500
Quantification of Inter-Sample Differences in T-Cell Receptor Repertoires Using Sequence-Based Information
Abstract
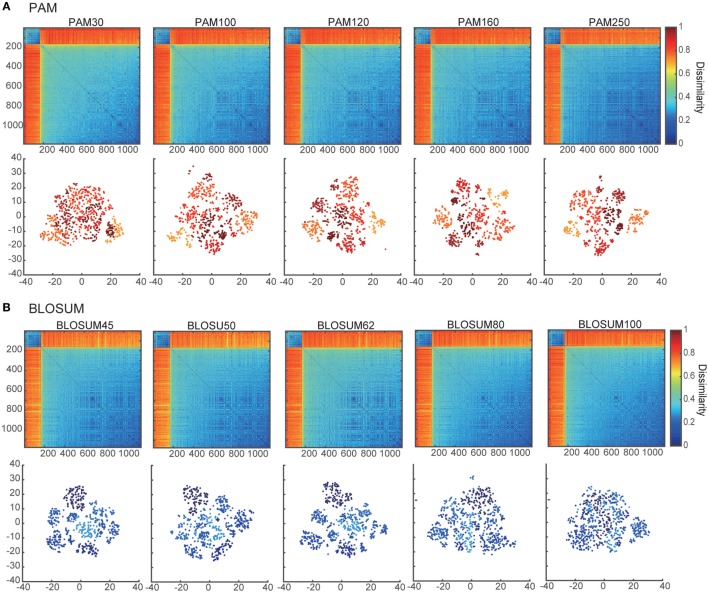
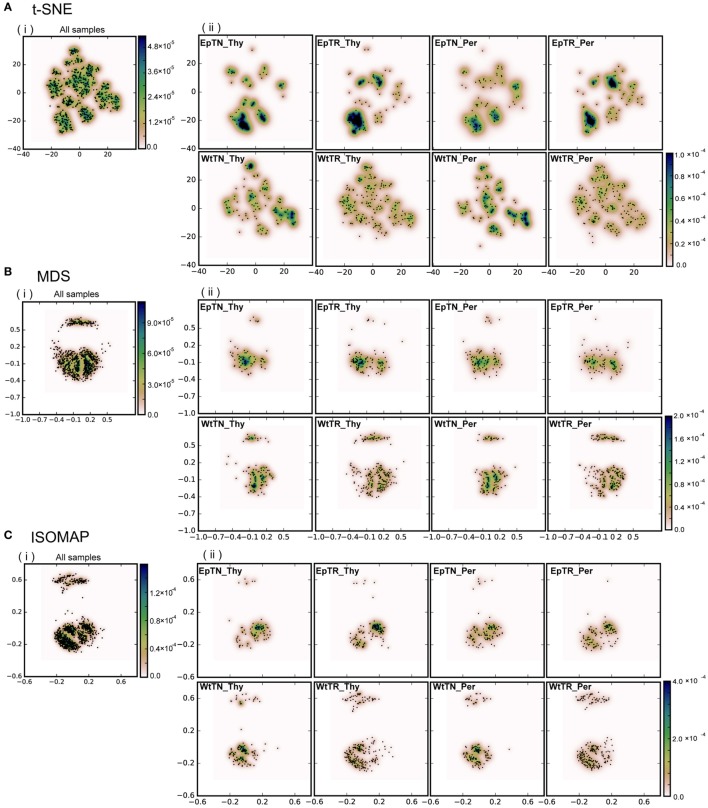
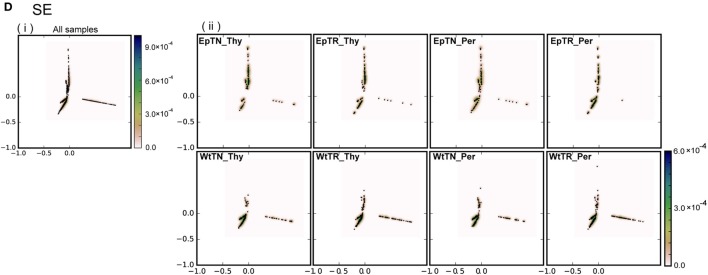
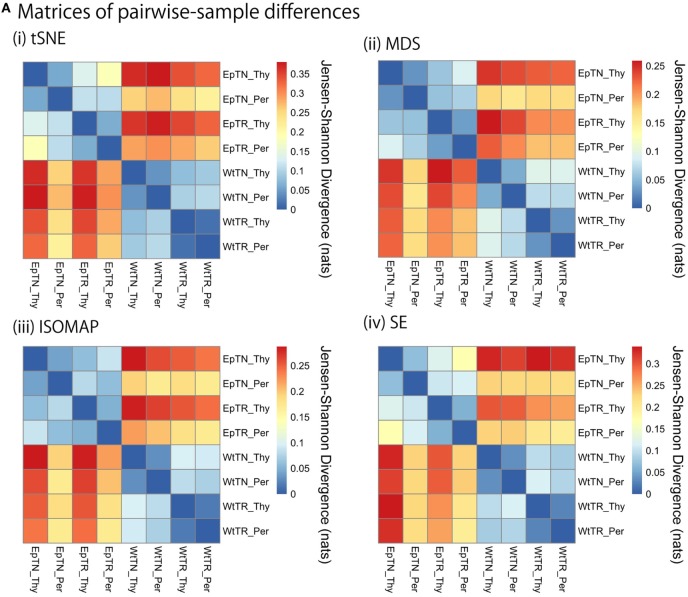
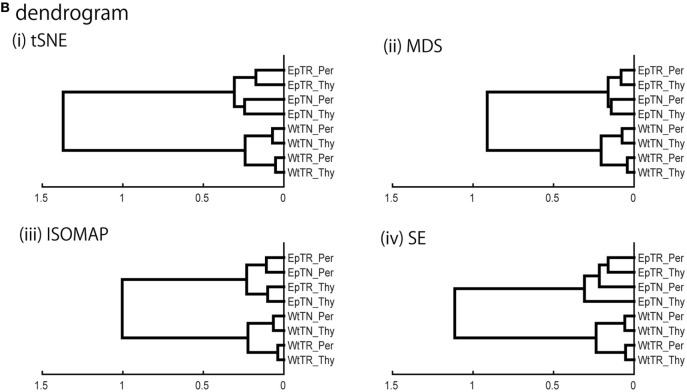
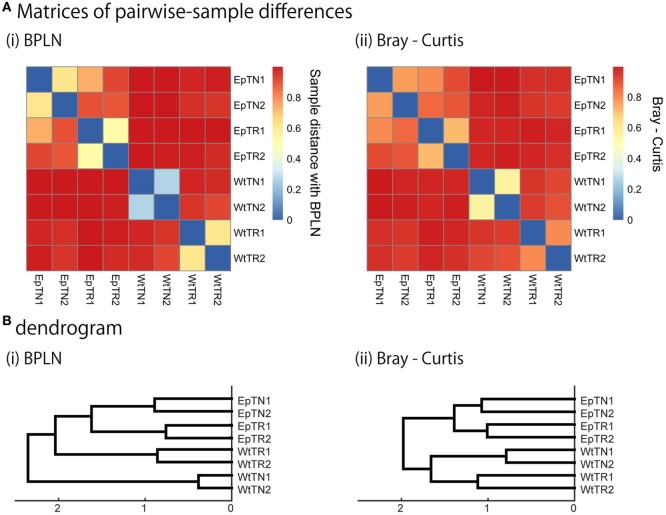
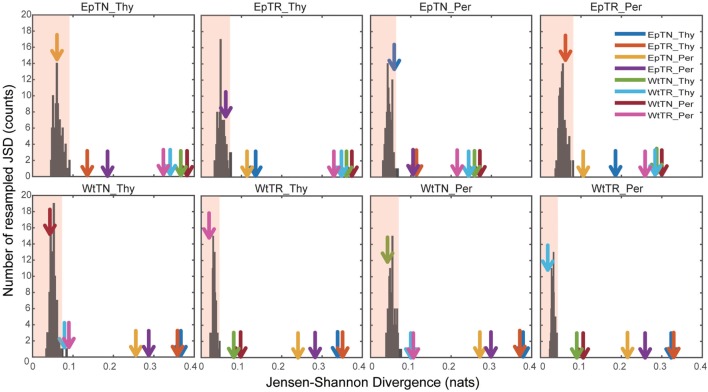
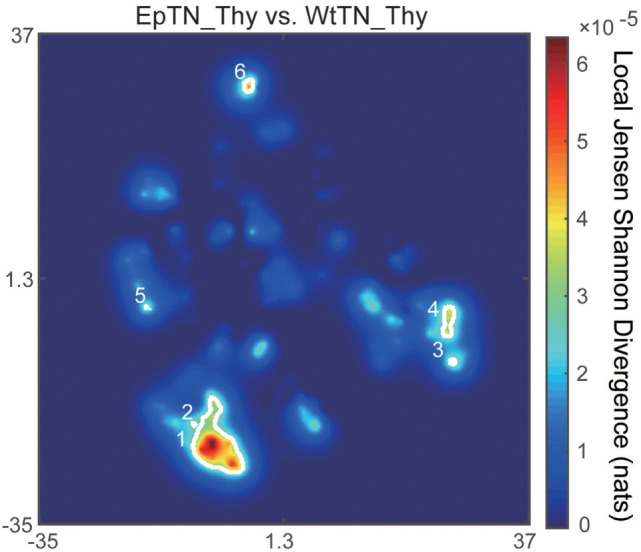
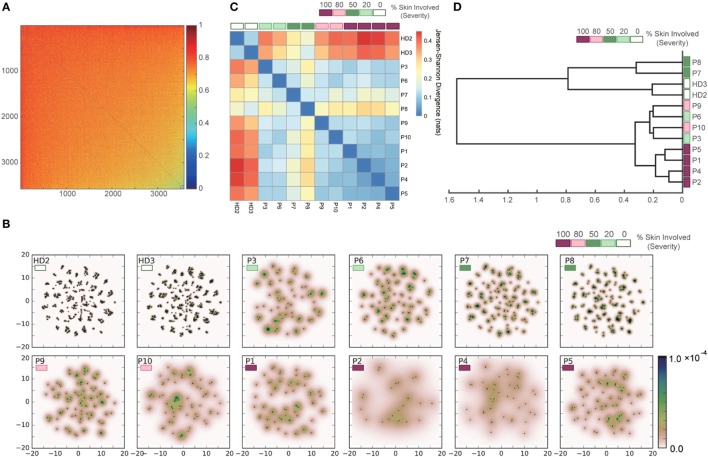
Inter-sample comparisons of T-cell receptor (TCR) repertoires are crucial for gaining a better understanding of the immunological states determined by different collections of T cells from different donor sites, cell types, and genetic and pathological backgrounds. For quantitative comparison, most previous studies utilized conventional methods in ecology, which focus on TCR sequences that overlap between pairwise samples. Some recent studies attempted another approach that is categorized into Poisson abundance models using the abundance distribution of observed TCR sequences. However, these methods ignore the details of the measured sequences and are consequently unable to identify sub-repertoires that might have important contributions to the observed inter-sample differences. Moreover, the sparsity of sequence data due to the huge diversity of repertoires hampers the performance of these methods, especially when few overlapping sequences exist. In this paper, we propose a new approach for REpertoire COmparison in Low Dimensions (RECOLD) based on TCR sequence information, which can estimate the low-dimensional structure by embedding the pairwise sequence dissimilarities in high-dimensional sequence space. The inter-sample differences between repertoires are then quantified by information-theoretic measures among the distributions of data estimated in the embedded space. Using datasets of mouse and human TCR repertoires, we demonstrate that RECOLD can accurately identify the inter-sample hierarchical structures, which have a good correspondence with our intuitive understanding about sample conditions. Moreover, for the dataset of transgenic mice that have strong restrictions on the diversity of their repertoires, our estimated inter-sample structure was consistent with the structure estimated by previous methods based on abundance or overlapping sequence information. For the dataset of human healthy donors and Sézary syndrome patients, our method also showed robust estimation performance even under the condition of high sparsity in TCR sequences, while previous studies failed to estimate the structure. In addition, we identified the sequences that contribute to the pairwise-sample differences between the repertoires with the different genetic backgrounds of mice. Such identification of the sequences contributing to variation in immune cell repertoires may provide substantial insight for the development of new immunotherapies and vaccines.
Keywords: Jensen–Shannon divergence; T cell; TCR repertoire; inter-repertoire comparison; manifold learning; pairwise sequence alignment; sequence dissimilarity.
Figures

Similar articles
-
Estimation of T-cell repertoire diversity and clonal size distribution by Poisson abundance models.J Immunol Methods. 2010 Feb 28;353(1-2):124-37. doi: 10.1016/j.jim.2009.11.009. Epub 2009 Nov 18. J Immunol Methods. 2010. PMID: 19931272
-
Clustering based approach for population level identification of condition-associated T-cell receptor β-chain CDR3 sequences.BMC Bioinformatics. 2021 Mar 25;22(1):159. doi: 10.1186/s12859-021-04087-7. BMC Bioinformatics. 2021. PMID: 33765908 Free PMC article.
-
Characterization of human T cell receptor repertoire data in eight thymus samples and four related blood samples.Data Brief. 2021 Jan 20;35:106751. doi: 10.1016/j.dib.2021.106751. eCollection 2021 Apr. Data Brief. 2021. PMID: 33553521 Free PMC article.
-
Sequence analysis of T-cell repertoires in health and disease.Genome Med. 2013 Oct 30;5(10):98. doi: 10.1186/gm502. eCollection 2013. Genome Med. 2013. PMID: 24172704 Free PMC article. Review.
-
New perspectives for large-scale repertoire analysis of immune receptors.Mol Immunol. 2008 May;45(9):2437-45. doi: 10.1016/j.molimm.2007.12.018. Epub 2008 Feb 14. Mol Immunol. 2008. PMID: 18279958 Review.
Cited by
-
Computational Strategies for Dissecting the High-Dimensional Complexity of Adaptive Immune Repertoires.Front Immunol. 2018 Feb 21;9:224. doi: 10.3389/fimmu.2018.00224. eCollection 2018. Front Immunol. 2018. PMID: 29515569 Free PMC article. Review.
-
Deep learning-based prediction of autoimmune diseases.Sci Rep. 2025 Feb 7;15(1):4576. doi: 10.1038/s41598-025-88477-4. Sci Rep. 2025. PMID: 39920178 Free PMC article.
-
Methods for sequence and structural analysis of B and T cell receptor repertoires.Comput Struct Biotechnol J. 2020 Jul 17;18:2000-2011. doi: 10.1016/j.csbj.2020.07.008. eCollection 2020. Comput Struct Biotechnol J. 2020. PMID: 32802272 Free PMC article. Review.
-
The Bayesian optimist's guide to adaptive immune receptor repertoire analysis.Immunol Rev. 2018 Jul;284(1):148-166. doi: 10.1111/imr.12664. Immunol Rev. 2018. PMID: 29944760 Free PMC article. Review.
-
Spatiotemporal Single-Cell Analysis Reveals T Cell Clonal Dynamics and Phenotypic Plasticity in Human Graft-versus-Host Disease.bioRxiv [Preprint]. 2025 May 28:2025.05.24.655962. doi: 10.1101/2025.05.24.655962. bioRxiv. 2025. PMID: 40501545 Free PMC article. Preprint.
References
-
- Bray JR, Curtis JT. An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr (1957) 27(4):325–49.10.2307/1942268 - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases