

Follicular Stimulating Hormone Accelerates Atherogenesis by Increasing Endothelial VCAM-1 Expression
- PMID: 29187895
- PMCID: PMC5706091
- DOI: 10.7150/thno.21216
Follicular Stimulating Hormone Accelerates Atherogenesis by Increasing Endothelial VCAM-1 Expression
Abstract
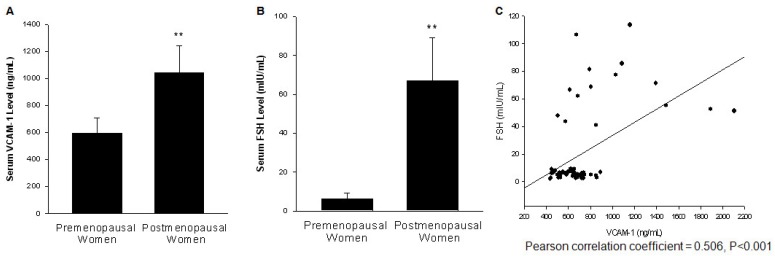
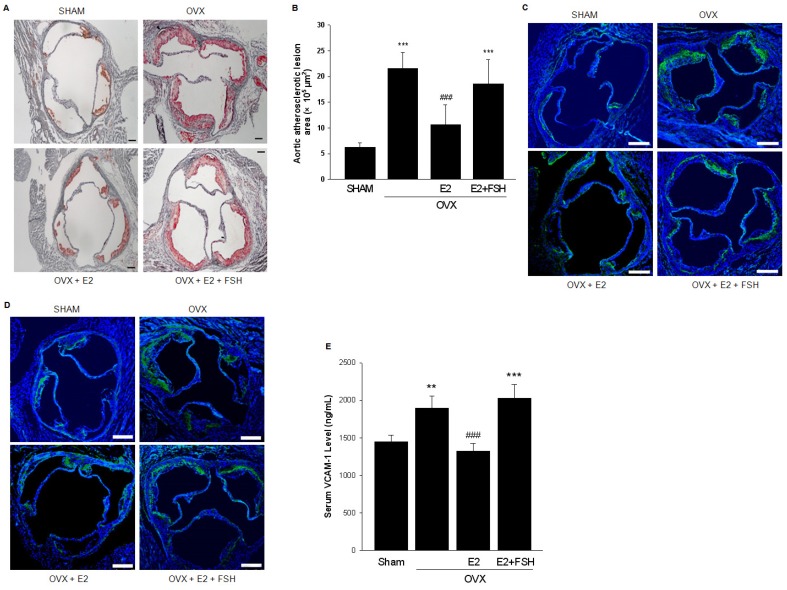
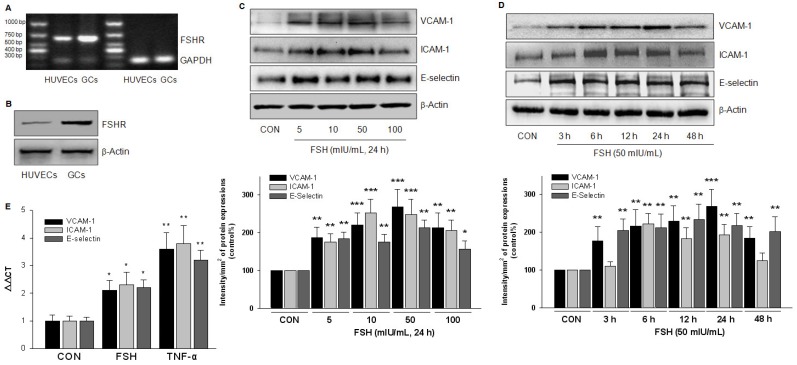
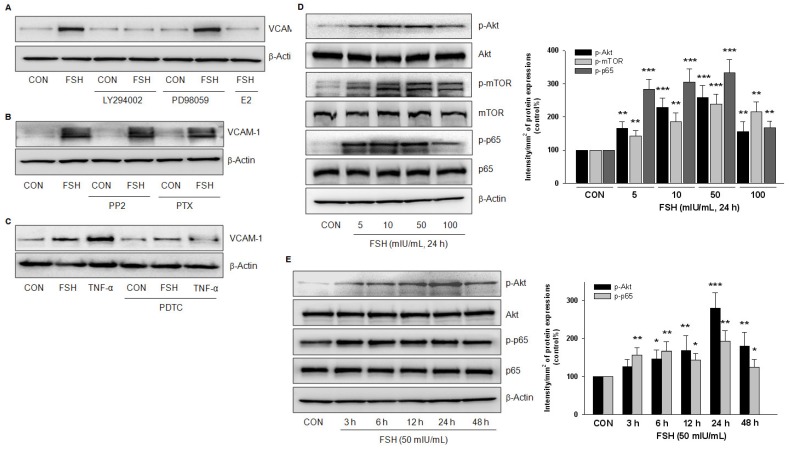
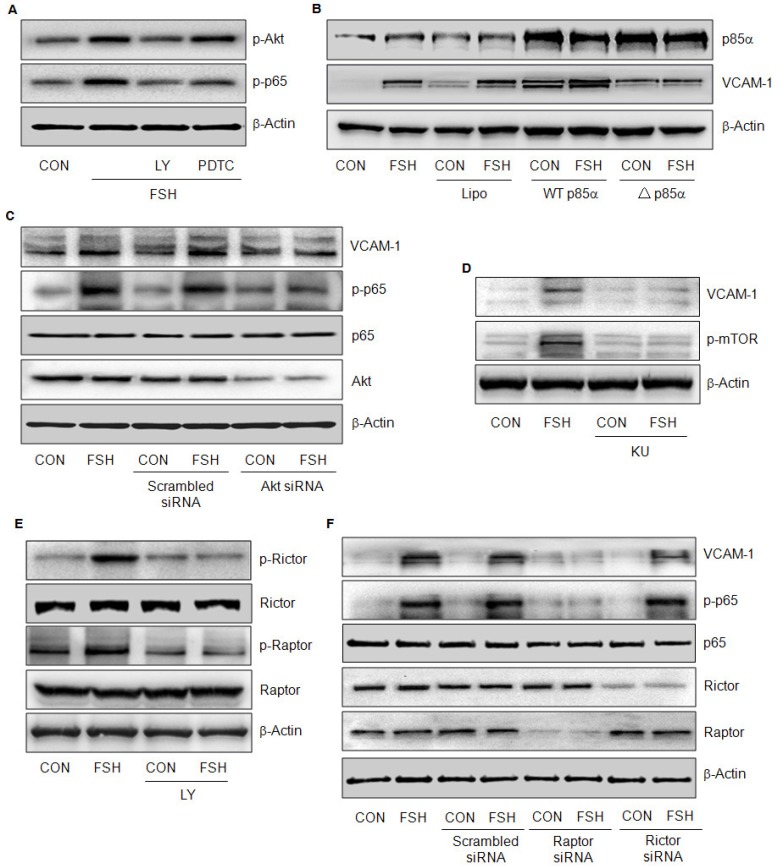
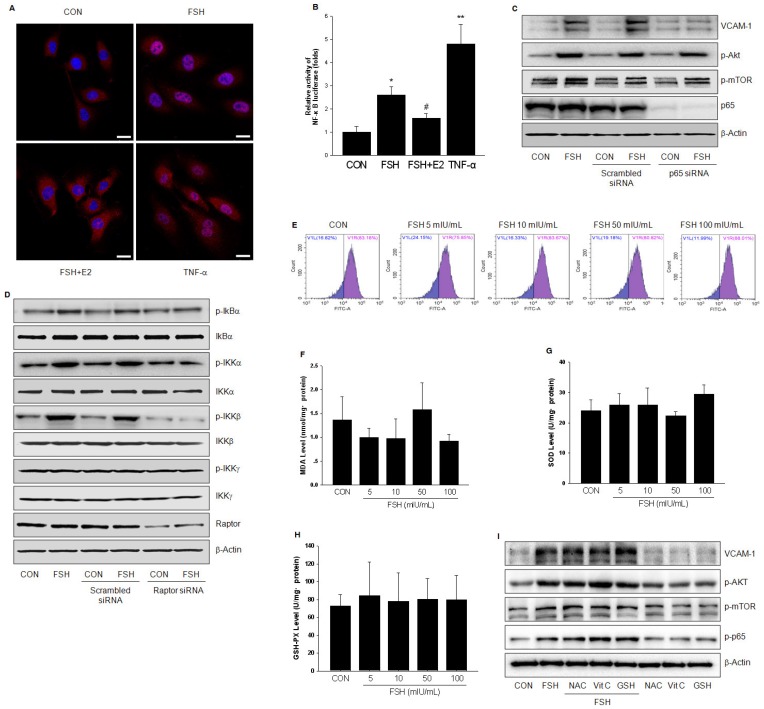
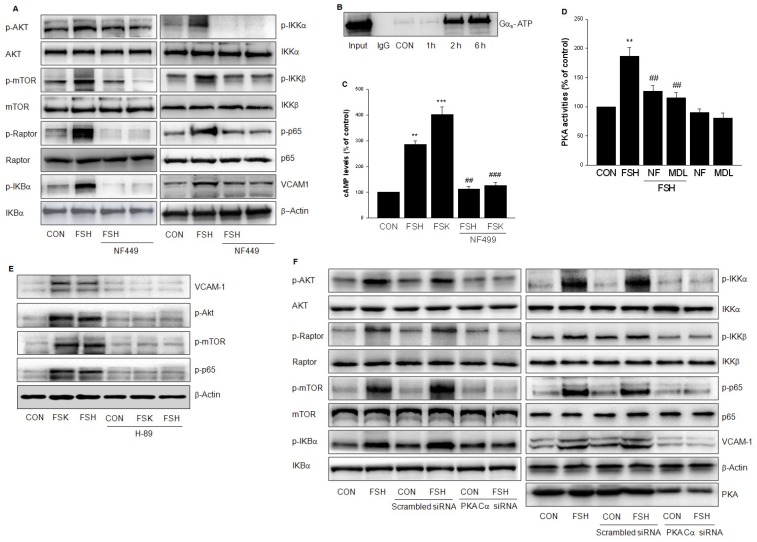
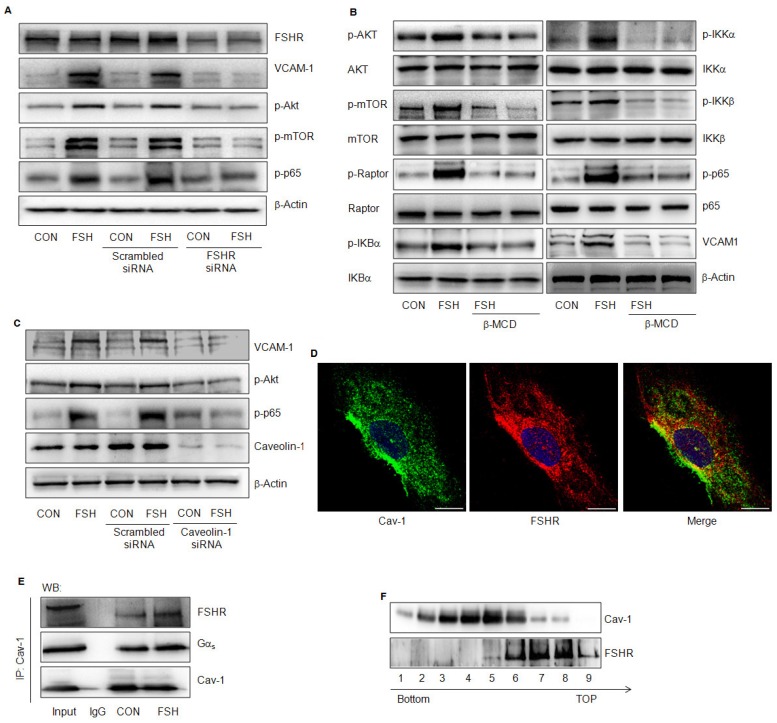
Rationale: Postmenopausal atherosclerosis (AS) has for decades been attributed to estrogen deficiency. Although the follicular stimulating hormone (FSH) levels rise sharply in parallel, the direct effect of FSH on AS has never been investigated. In this study, we explored the possible role of FSH in the development of AS. Methods: This was a prospective cohort study of 48 healthy premenopausal and 15 postmenopausal women. ApoE knockout mice were used as atherosclerosis model and human umbilical vascular endothelial cells (HUVECs) were cultured as cell model. Serum hormones and vascular cell adhesion molecule-1 (VCAM-1) levels were measured. Real-time PCR, histology for atherosclerotic lesions, immunofluorescence, luciferase assay, transfection experiments, flow chamber adhesion assay and western blot were performed. Results: In ApoE knockout mice, administration of FSH increased the atherosclerotic lesions and serum VCAM-1 concentration. Importantly, in blood samples of postmenopausal women, we detected significantly higher levels of FSH and VCAM-1 compared with those from premenopausal women, and there was a positive correlation between these two molecules. In cultured HUVECs, FSH receptor (FSHR) mRNA and protein expression were detected and FSH enhanced VCAM-1 expression. This effect was mediated by the activation of nuclear factor κB (NF-κB), which was sequentially enhanced by the activation of PI3K/Akt/mTOR cascade. FSH first enhanced GαS activity resulting in elevated cAMP level and PKA activity, which relayed the signals from FSHR to the PI3K/Akt/mTOR cascade. Furthermore, FSHR was detected in endothelial caveolae fraction and interacted with caveolin-1 and GαS. The disruption of caveolae or the silencing of caveolin-1 blocked FSH effects on signaling activation and VCAM-1 expression, suggesting the existence of a functional signaling module in membrane caveolae. Finally, FSH increased human monocyte adhesion to HUVECs which was reversed by the VCAM-1 neutralizing antibody. Conclusion: FSHR was located in the membrane caveolae of HUVECs and FSH promoted VCAM-1 expression via FSHR/GαS /cAMP/PKA and PI3K/Akt/mTOR/NF-κB pathway. This may contribute to the deleterious role of FSH in the development of AS in postmenopausal women.
Keywords: Atherosclerosis; Caveolae.; Follicular Stimulating Hormone; Vascular Cell Adhesion Molecule-1; Vascular Endothelial Cells.
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
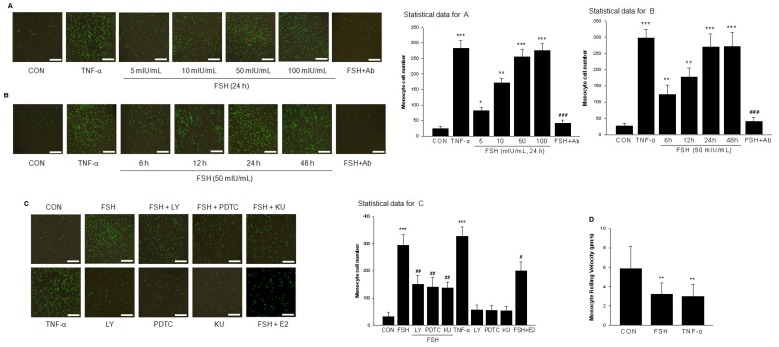
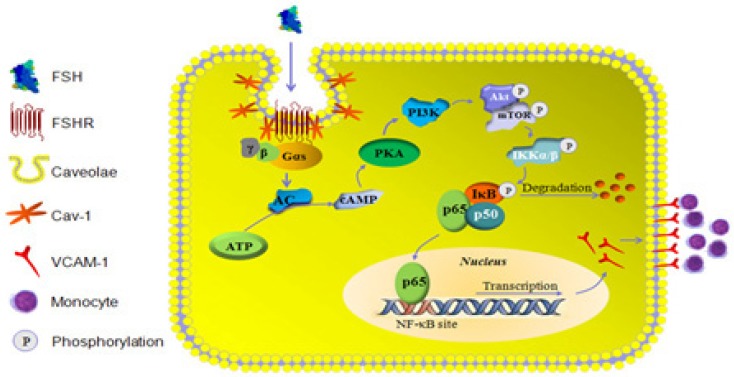
References
-
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M. et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–360. - PubMed
-
- Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–11. - PubMed
-
- Harman SM. Menopausal hormone treatment cardiovascular disease: another look at an unresolved conundrum. Fertil Steril. 2014;101:887–97. - PubMed
-
- Genazzani AR, Simoncini T. Pharmacotherapy: Benefits of menopausal hormone therapy-timing is key. Nat Rev Endocrinol. 2013;9:5–6. - PubMed
-
- Randolph JF Jr, Sowers M, Bondarenko IV, Harlow SD, Luborsky JL, Little RJ. Change in estradiol and follicle-stimulating hormone across the early menopausal transition: effects of ethnicity and age. J Clin Endocrinol Metab. 2004;89:1555–61. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
