

Novel Dominant-Negative GH Receptor Mutations Expands the Spectrum of GHI and IGF-I Deficiency
- PMID: 29188236
- PMCID: PMC5686656
- DOI: 10.1210/js.2016-1119
Novel Dominant-Negative GH Receptor Mutations Expands the Spectrum of GHI and IGF-I Deficiency
Abstract
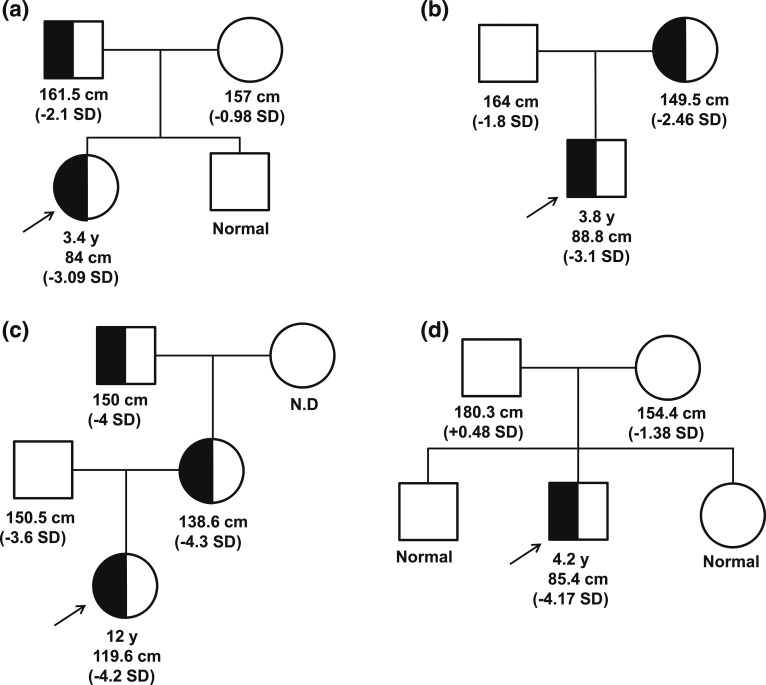
Context: Autosomal-recessive mutations in the growth hormone receptor (GHR) are the most common causes for primary growth hormone insensitivity (GHI) syndrome with classical GHI phenotypically characterized by severe short stature and marked insulin-like growth factor (IGF)-I deficiency. We report three families with dominant-negative heterozygous mutations in the intracellular domain of the GHR causing a nonclassical GHI phenotype.
Objective: To determine if the identified GHR heterozygous variants exert potential dominant-negative effects and are the cause for the GHI phenotype in our patients.
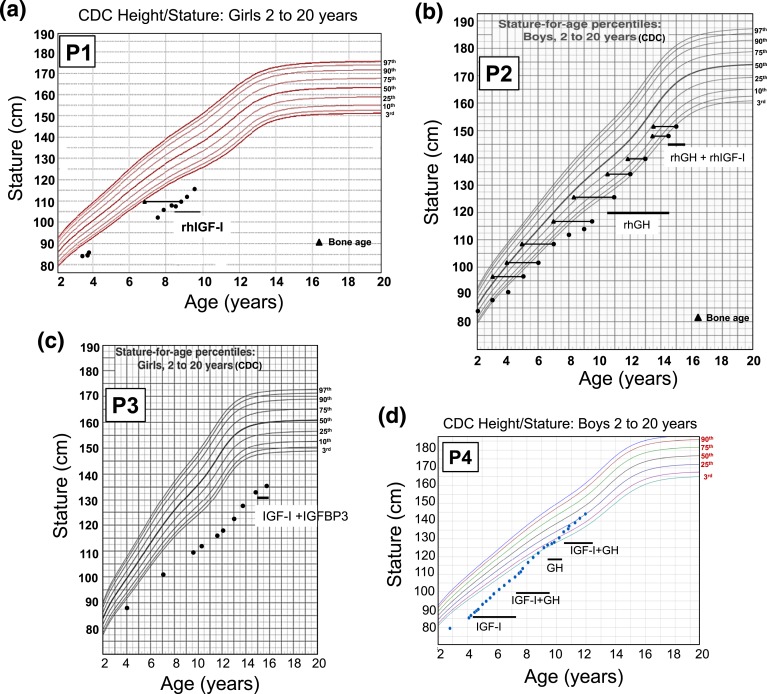
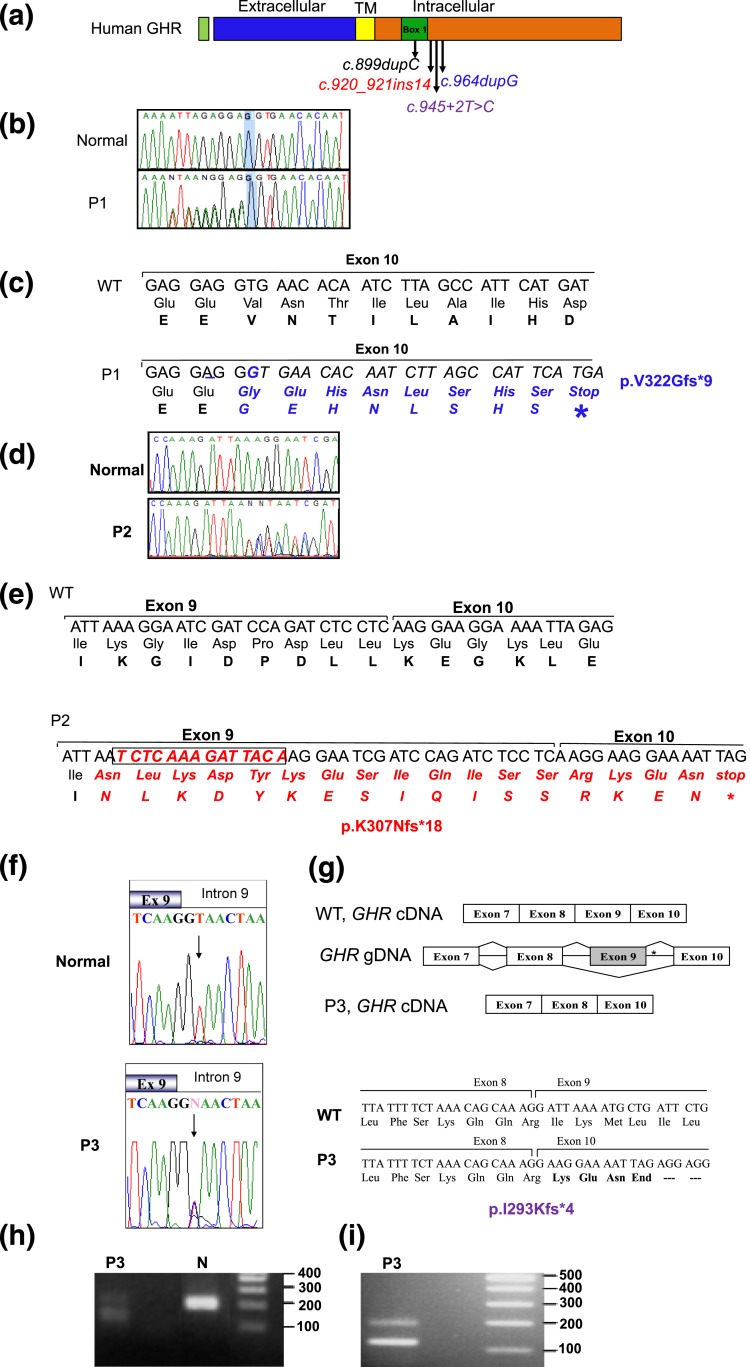
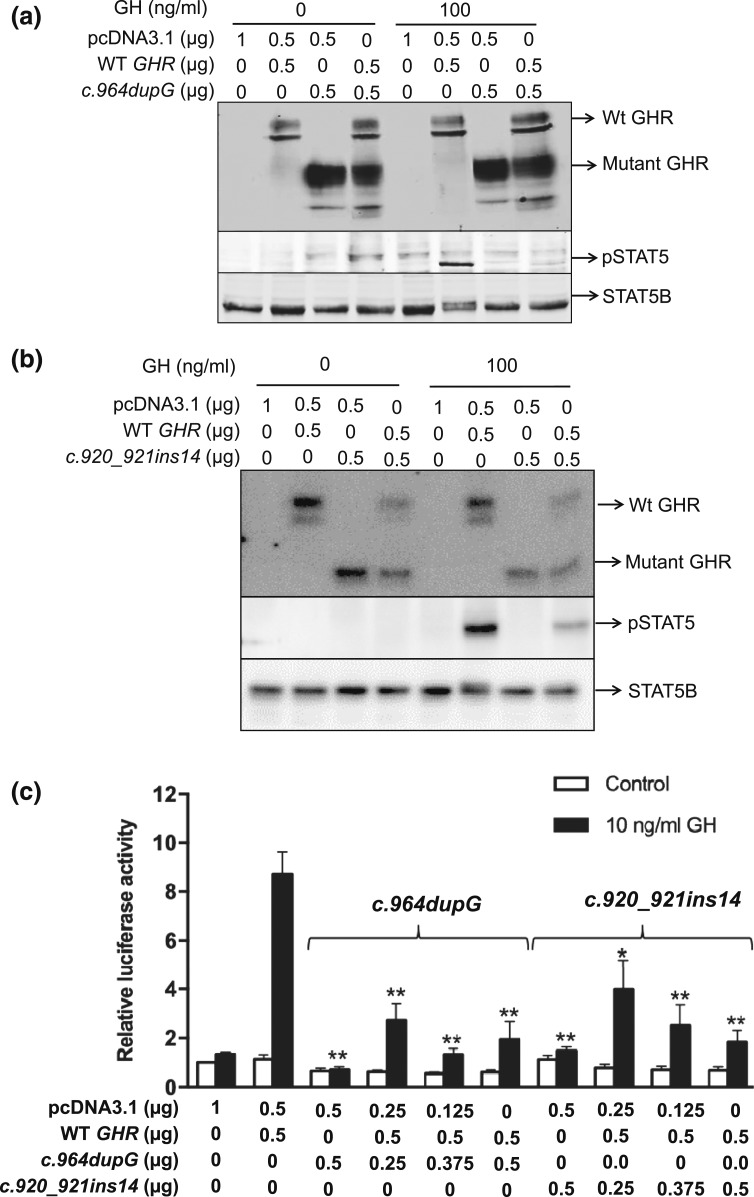
Results: All three mutations (c.964dupG, c.920_921insTCTCAAAGATTACA, and c.945+2T>C) are predicted to result in frameshift and early protein termination. In vitro functional analysis of variants c.964dupG and c.920_921insTCTCAAAGATTACA (c.920_921ins14) suggests that these variants are expressed as truncated proteins and, when coexpressed with wild-type GHR, mimicking the heterozygous state in our patients, exert dominant-negative effects. Additionally, we provide evidence that a combination therapy of recombinant human growth hormone (rhGH) and rhIGF-I improved linear growth to within normal range for one of our previously reported patients with a characterized, dominant-negative GHR (c.899dupC) mutation.
Conclusion: Dominant-negative GHR mutations are causal of the mild GHI with substantial growth failure observed in our patients. Heterozygous defects in the intracellular domain of GHR should, therefore, be considered in cases of idiopathic short stature and IGF-I deficiency. Combination therapy of rhGH and rhIGF-I improved growth in one of our patients.
Keywords: GH/IGF-I therapy; dominant-negative GHR mutations.
Figures
References
-
- Milward A, Metherell L, Maamra M, Barahona MJ, Wilkinson IR, Camacho-Hübner C, Savage MO, Bidlingmaier M, Clark AJ, Ross RJ, Webb SM. Growth hormone (GH) insensitivity syndrome due to a GH receptor truncated after Box1, resulting in isolated failure of STAT 5 signal transduction. J Clin Endocrinol Metab [published correction appears in J Clin Endocrinol Metab 2009;94(7):2674]. 2004;89(3):1259–1266. - PubMed
-
- Davey HW, Xie T, McLachlan MJ, Wilkins RJ, Waxman DJ, Grattan DR. STAT5b is required for GH-induced liver IGF-I gene expression. Endocrinology. 2001;142(9):3836–3841. - PubMed
-
- Woelfle J, Billiard J, Rotwein P. Acute control of insulin-like growth factor-I gene transcription by growth hormone through Stat5b. J Biol Chem. 2003;278(25):22696–22702. - PubMed
-
- Ooi GT, Cohen FJ, Tseng LY, Rechler MM, Boisclair YR. Growth hormone stimulates transcription of the gene encoding the acid-labile subunit (ALS) of the circulating insulin-like growth factor-binding protein complex and ALS promoter activity in rat liver. Mol Endocrinol. 1997;11(7):997–1007. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources