

Efficient in situ barcode sequencing using padlock probe-based BaristaSeq
- PMID: 29190363
- PMCID: PMC5829746
- DOI: 10.1093/nar/gkx1206
Efficient in situ barcode sequencing using padlock probe-based BaristaSeq
Abstract
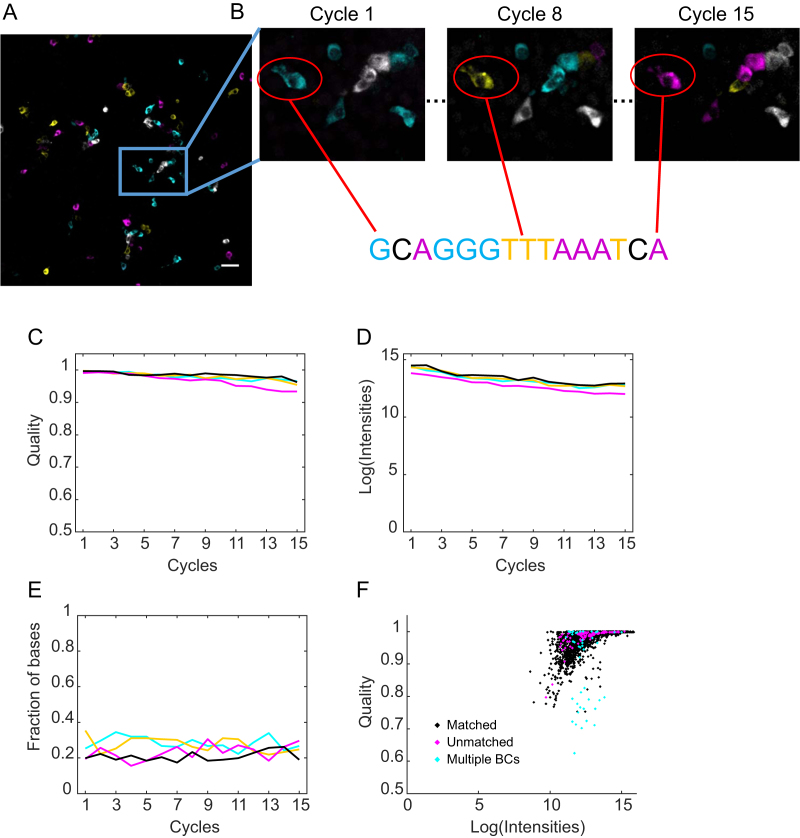
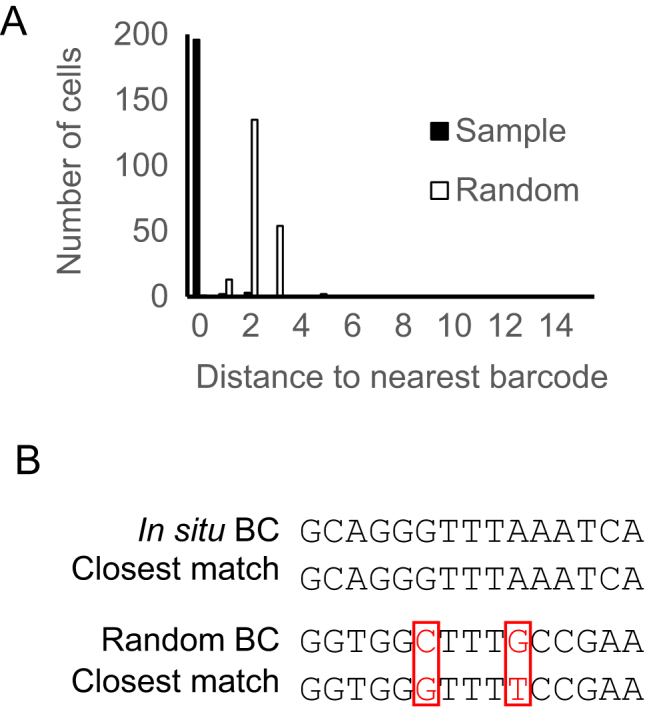
Cellular DNA/RNA tags (barcodes) allow for multiplexed cell lineage tracing and neuronal projection mapping with cellular resolution. Conventional approaches to reading out cellular barcodes trade off spatial resolution with throughput. Bulk sequencing achieves high throughput but sacrifices spatial resolution, whereas manual cell picking has low throughput. In situ sequencing could potentially achieve both high spatial resolution and high throughput, but current in situ sequencing techniques are inefficient at reading out cellular barcodes. Here we describe BaristaSeq, an optimization of a targeted, padlock probe-based technique for in situ barcode sequencing compatible with Illumina sequencing chemistry. BaristaSeq results in a five-fold increase in amplification efficiency, with a sequencing accuracy of at least 97%. BaristaSeq could be used for barcode-assisted lineage tracing, and to map long-range neuronal projections.
Figures
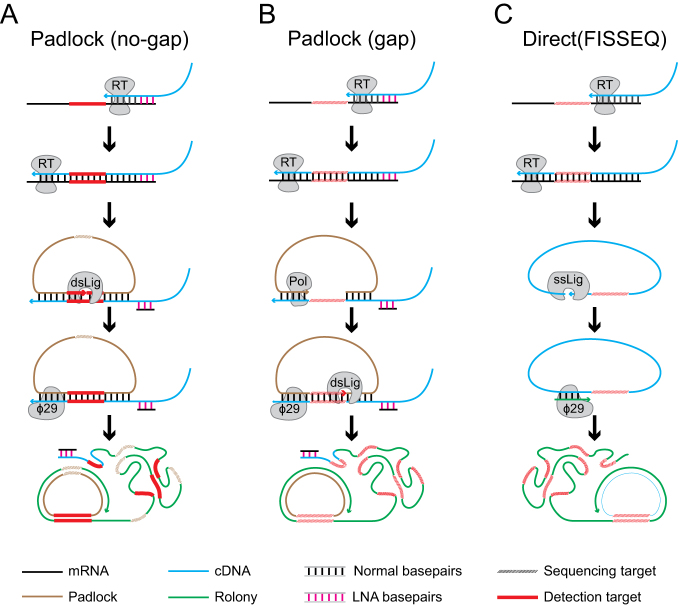
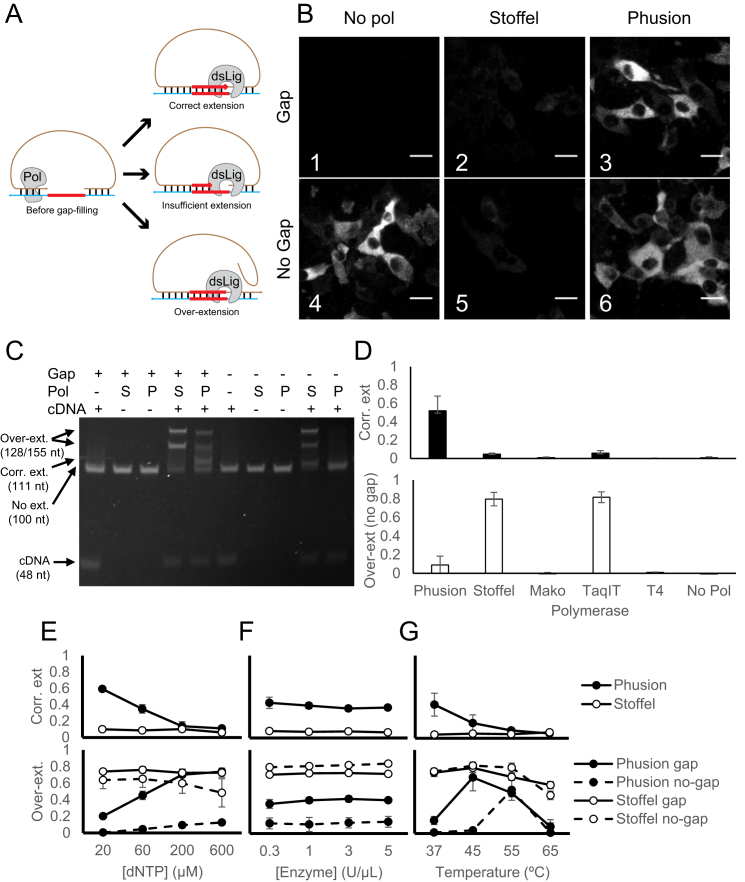
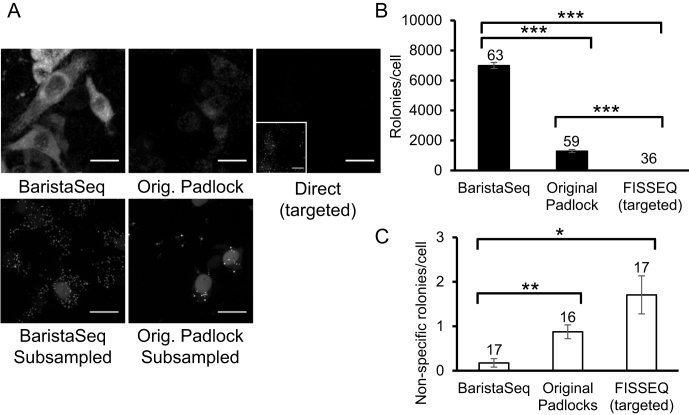
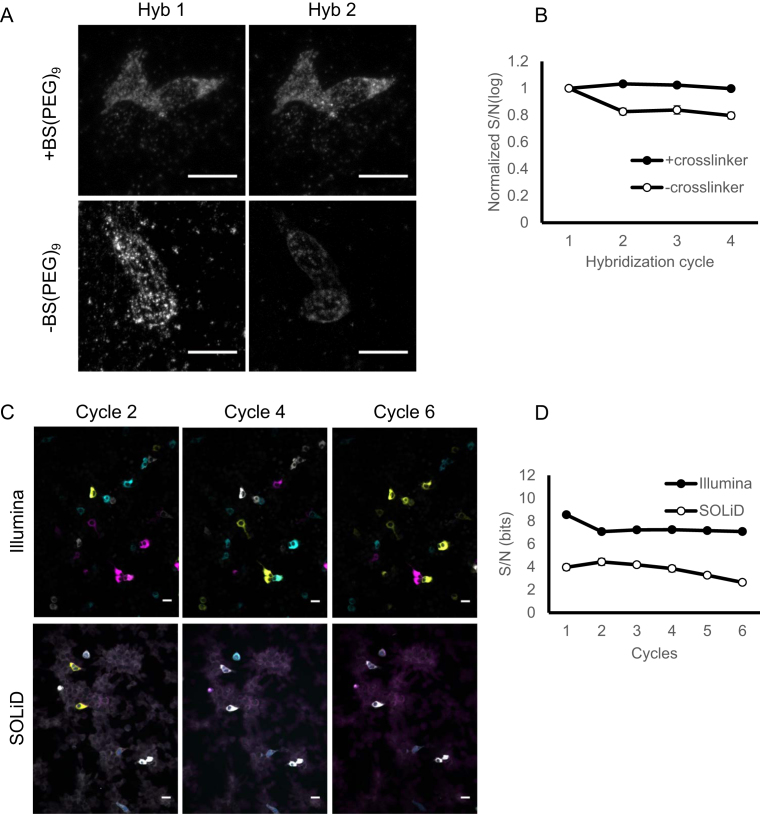
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
