

Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map
- PMID: 29190685
- PMCID: PMC5726721
- DOI: 10.1371/journal.pbio.2003213
Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map
Abstract
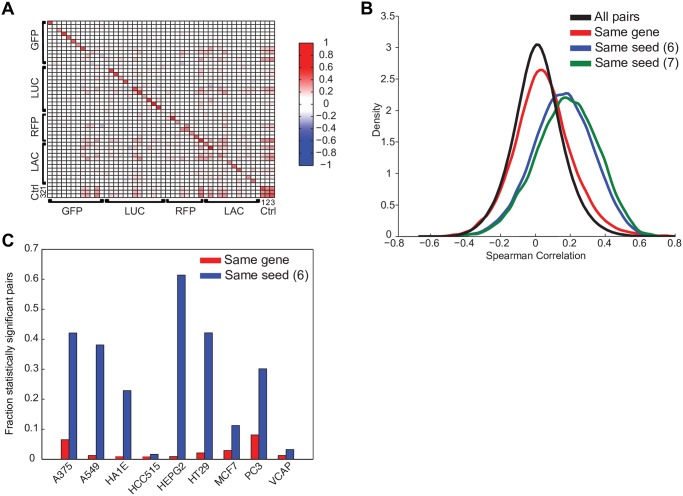
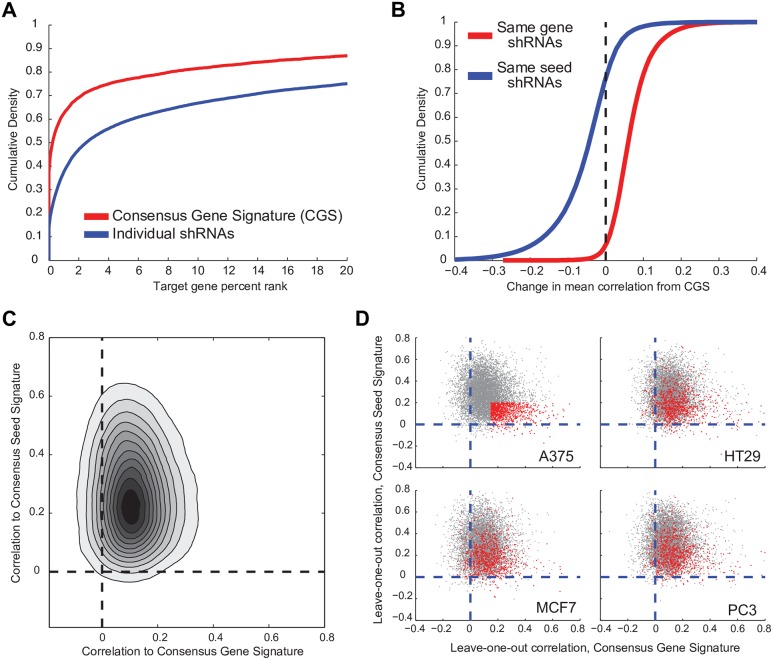
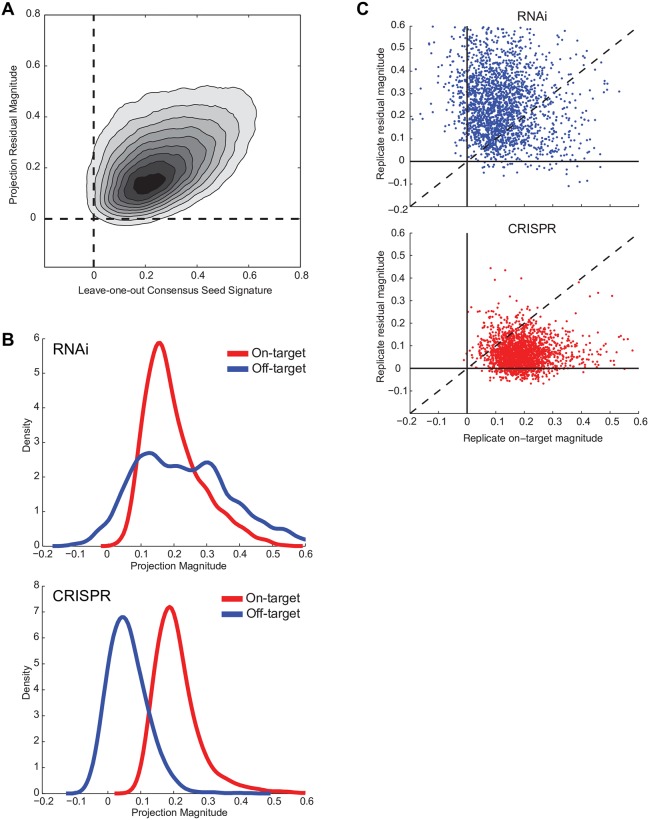
The application of RNA interference (RNAi) to mammalian cells has provided the means to perform phenotypic screens to determine the functions of genes. Although RNAi has revolutionized loss-of-function genetic experiments, it has been difficult to systematically assess the prevalence and consequences of off-target effects. The Connectivity Map (CMAP) represents an unprecedented resource to study the gene expression consequences of expressing short hairpin RNAs (shRNAs). Analysis of signatures for over 13,000 shRNAs applied in 9 cell lines revealed that microRNA (miRNA)-like off-target effects of RNAi are far stronger and more pervasive than generally appreciated. We show that mitigating off-target effects is feasible in these datasets via computational methodologies to produce a consensus gene signature (CGS). In addition, we compared RNAi technology to clustered regularly interspaced short palindromic repeat (CRISPR)-based knockout by analysis of 373 single guide RNAs (sgRNAs) in 6 cells lines and show that the on-target efficacies are comparable, but CRISPR technology is far less susceptible to systematic off-target effects. These results will help guide the proper use and analysis of loss-of-function reagents for the determination of gene function.
Conflict of interest statement
We have read the journal's policy and the authors of this manuscript have the following competing interests: A.S. is a shareholder of Genometry, Inc.
Figures



References
-
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. American Association for the Advancement of Science; 2006;313: 1929–1935. doi: 10.1126/science.1132939 - DOI - PubMed
-
- Belcastro V, Siciliano V, Gregoretti F, Mithbaokar P, Dharmalingam G, Berlingieri S, et al. Transcriptional gene network inference from a massive dataset elucidates transcriptome organization and gene function. Nucleic Acids Research. 2011;39: 8677–8688. doi: 10.1093/nar/gkr593 - DOI - PMC - PubMed
-
- Wang Z, Monteiro CD, Jagodnik KM, Fernandez NF, Gundersen GW, Rouillard AD, et al. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nature Communications. Nature Publishing Group; 2016;7: 12846 doi: 10.1038/ncomms12846 - DOI - PMC - PubMed
-
- Pirhaji L, Milani P, Leidl M, Curran T, Avila-Pacheco J, Clish CB, et al. Revealing disease-associated pathways by network integration of untargeted metabolomics. Nature Methods. Nature Research; 2016;13: 770–776. doi: 10.1038/nmeth.3940 - DOI - PMC - PubMed
-
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. A Next Generation Connectivity Map: L1000 Platform And The First 1,000,000 Profiles. bioRxiv. Cold Spring Harbor Labs Journals; 2017;: 136168 doi: 10.1101/136168 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
