

Laminin-deficient muscular dystrophy: Molecular pathogenesis and structural repair strategies
- PMID: 29191403
- PMCID: PMC5971131
- DOI: 10.1016/j.matbio.2017.11.009
Laminin-deficient muscular dystrophy: Molecular pathogenesis and structural repair strategies
Abstract
Laminins are large heterotrimers composed of the α, β and γ subunits with distinct tissue-specific and developmentally regulated expression patterns. The laminin-α2 subunit, encoded by the LAMA2 gene, is expressed in skeletal muscle, Schwann cells of the peripheral nerve and astrocytes and pericytes of the capillaries in the brain. Mutations in LAMA2 cause the most common type of congenital muscular dystrophies, called LAMA2 MD or MDC1A. The disorder manifests mostly as a muscular dystrophy but slowing of nerve conduction contributes to the disease. There are severe, non-ambulatory or milder, ambulatory variants, the latter resulting from reduced laminin-α2 expression and/or deficient laminin-α2 function. Lm-211 (α2β1γ1) is responsible for initiating basement membrane assembly. This is primarily accomplished by anchorage of Lm-211 to dystroglycan and α7β1 integrin receptors, polymerization, and binding to nidogen and other structural components. In LAMA2 MD, Lm-411 replaces Lm-211; however, Lm-411 lacks the ability to polymerize and bind to receptors. This results in a weakened basement membrane leading to the disease. The possibility of introducing structural repair proteins that correct the underlying abnormality is an attractive therapeutic goal. Recent studies in mouse models for LAMA2 MD reveal that introduction of laminin-binding linker proteins that restore lost functional activities can substantially ameliorate the disease. This review discusses the underlying mechanism of this repair and compares this approach to other developing therapies employing pharmacological treatments.
Copyright © 2018 International Society of Matrix Biology. Published by Elsevier B.V. All rights reserved.
Figures
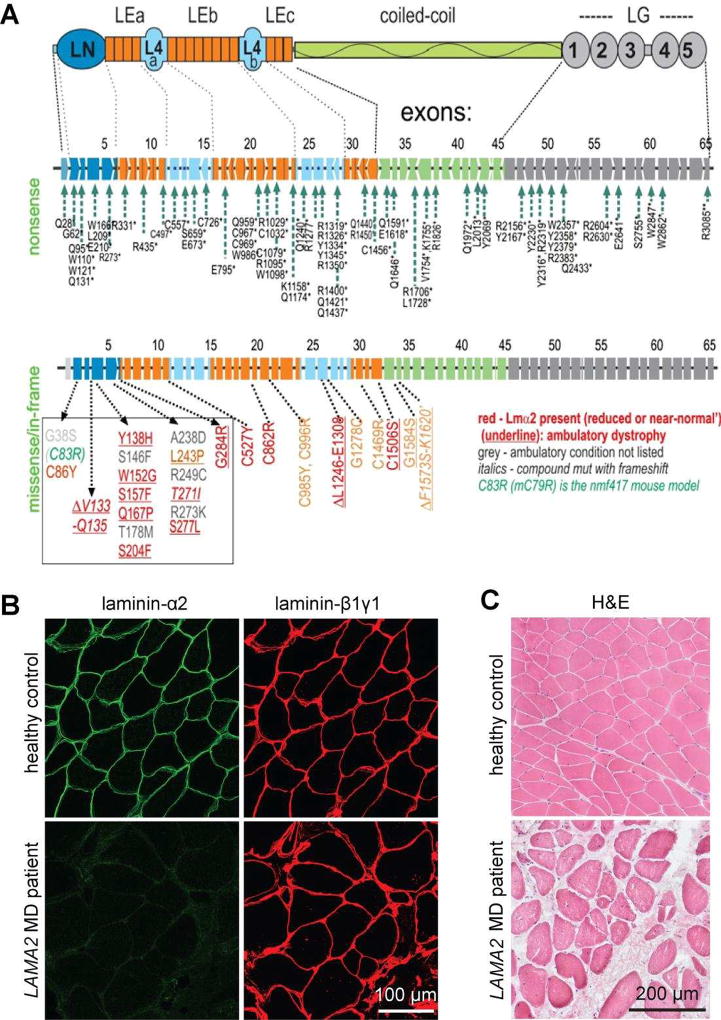
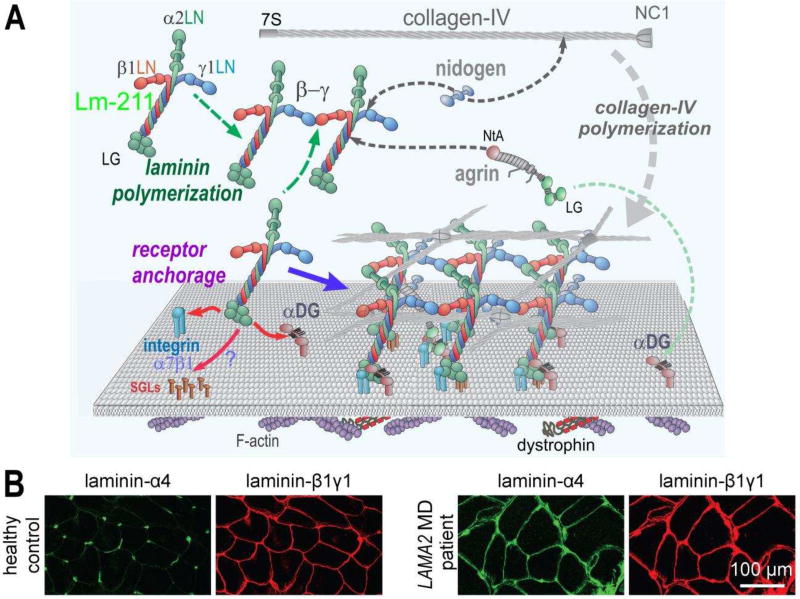
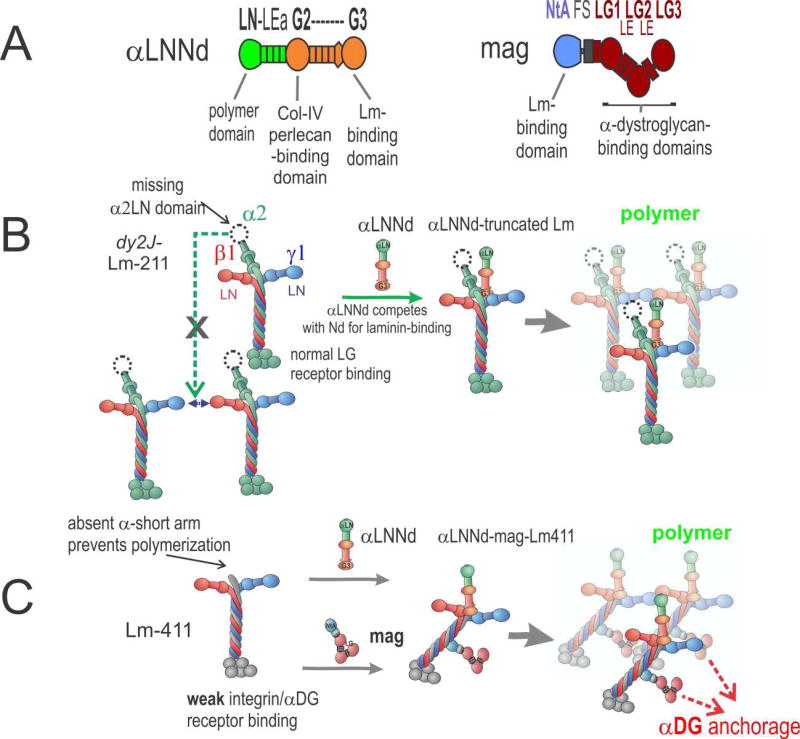
References
-
- Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JC, Kleinman HK, Marinkovich MP, Martin GR, Mayer U, Meneguzzi G, Miner JH, Miyazaki K, Patarroyo M, Paulsson M, Quaranta V, Sanes JR, Sasaki T, Sekiguchi K, Sorokin LM, Talts JF, Tryggvason K, Uitto J, Virtanen I, von der Mark K, Wewer UM, Yamada Y, Yurchenco PD. A simplified laminin nomenclature. Matrix Biol. 2005;24(5):326–32. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
