

TGF-β1/p53 signaling in renal fibrogenesis
- PMID: 29191563
- PMCID: PMC5860677
- DOI: 10.1016/j.cellsig.2017.11.005
TGF-β1/p53 signaling in renal fibrogenesis
Abstract
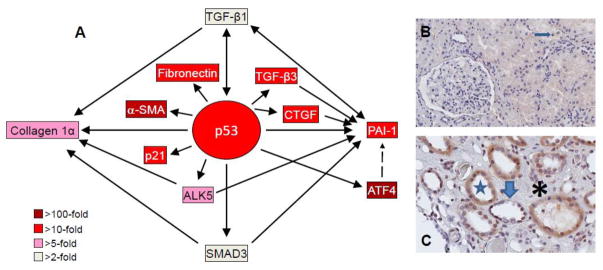
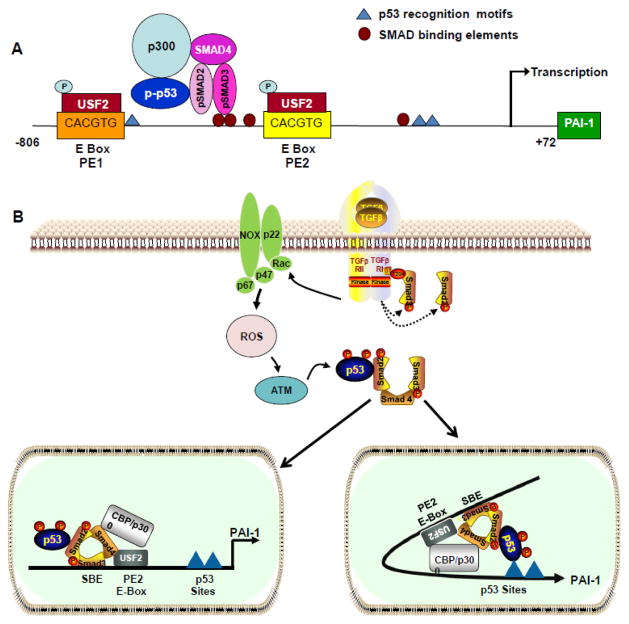
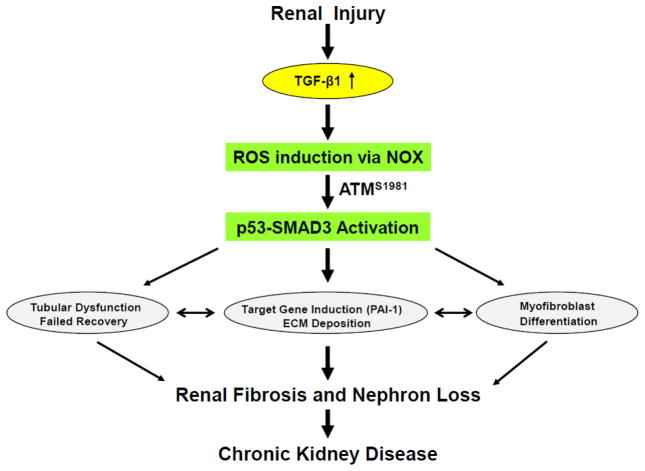
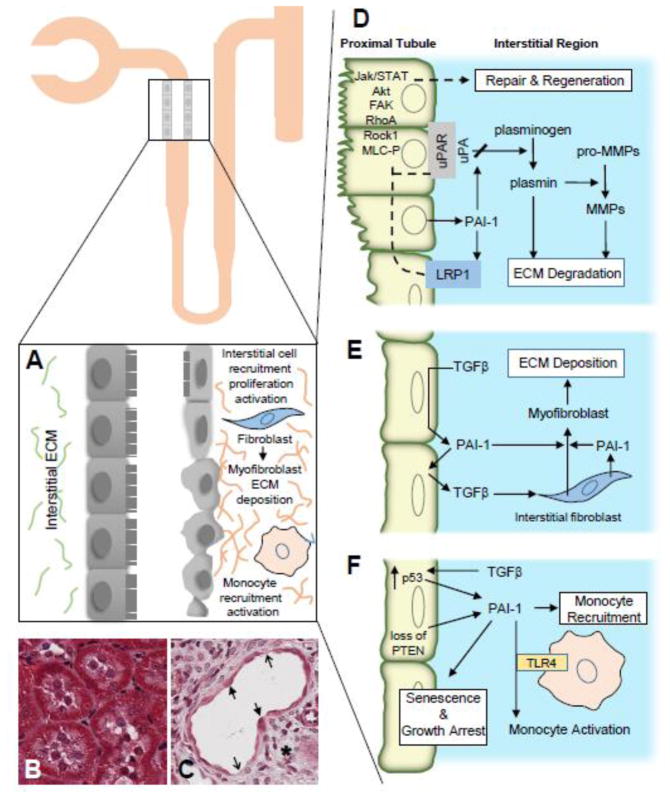
Fibrotic disorders of the renal, pulmonary, cardiac, and hepatic systems are associated with significant morbidity and mortality. Effective therapies to prevent or curtail the advancement to organ failure, however, remain a major clinical challenge. Chronic kidney disease, in particular, constitutes an increasing medical burden affecting >15% of the US population. Regardless of etiology (diabetes, hypertension, ischemia, acute injury, urologic obstruction), persistently elevated TGF-β1 levels are causatively linked to the activation of profibrotic signaling networks and disease progression. TGF-β1 is the principal driver of renal fibrogenesis, a dynamic pathophysiologic process that involves tubular cell injury/apoptosis, infiltration of inflammatory cells, interstitial fibroblast activation and excess extracellular matrix synthesis/deposition leading to impaired kidney function and, eventually, to chronic and end-stage disease. TGF-β1 activates the ALK5 type I receptor (which phosphorylates SMAD2/3) as well as non-canonical (e.g., src kinase, EGFR, JAK/STAT, p53) pathways that collectively drive the fibrotic genomic program. Such multiplexed signal integration has pathophysiological consequences. Indeed, TGF-β1 stimulates the activation and assembly of p53-SMAD3 complexes required for transcription of the renal fibrotic genes plasminogen activator inhibitor-1, connective tissue growth factor and TGF-β1. Tubular-specific ablation of p53 in mice or pifithrin-α-mediated inactivation of p53 prevents epithelial G2/M arrest, reduces the secretion of fibrotic effectors and attenuates the transition from acute to chronic renal injury, further supporting the involvement of p53 in disease progression. This review focuses on the pathophysiology of TGF-β1-initiated renal fibrogenesis and the role of p53 as a regulator of profibrotic gene expression.
Keywords: Fibrosis; Kidney; Plasminogen activator Inhibitor-1; TGF-β1; p53.
Copyright © 2017 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
References
-
- Zeisberg M, Neilson EG. Mechanisms of tubulointestinal fibrosis. J Am Soc Neprhol. 2010;21:181901834. - PubMed
-
- Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet. 2017;389:1238–1252. - PubMed
-
- Mei C, Zheng F. Chronic inflammation potentiates kidney aging. Semin Nephrol. 2009;29:555–568. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
