

The target landscape of clinical kinase drugs
- PMID: 29191878
- PMCID: PMC6542668
- DOI: 10.1126/science.aan4368
The target landscape of clinical kinase drugs
Abstract
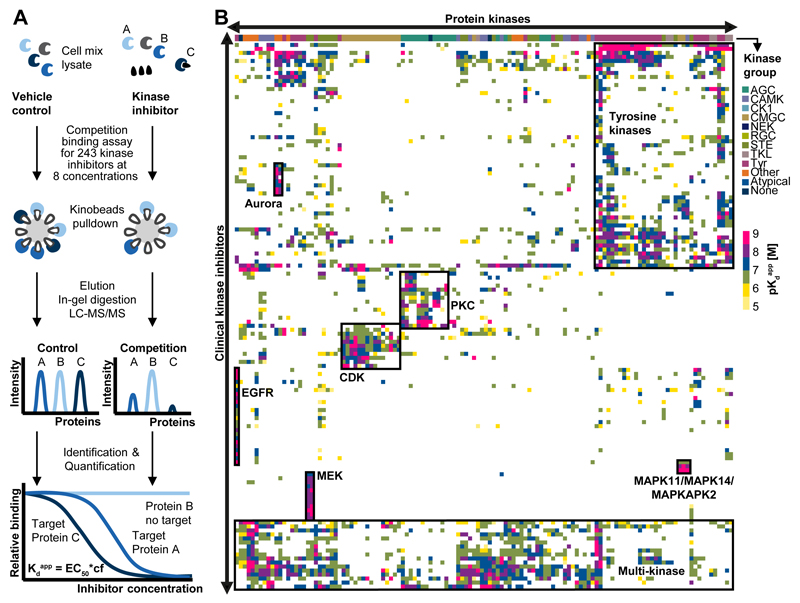
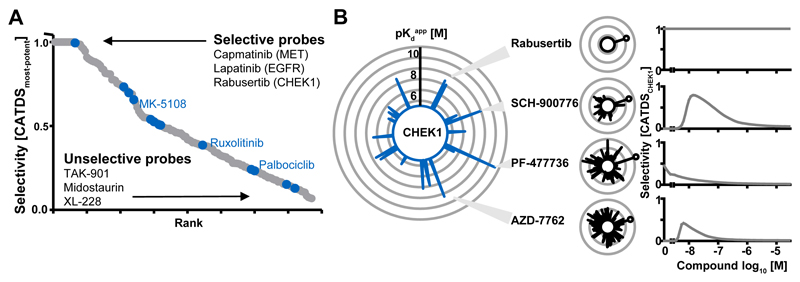
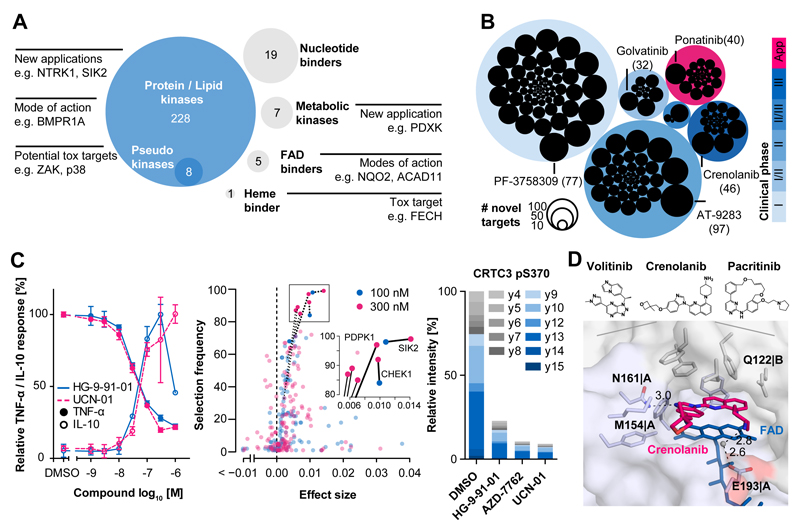
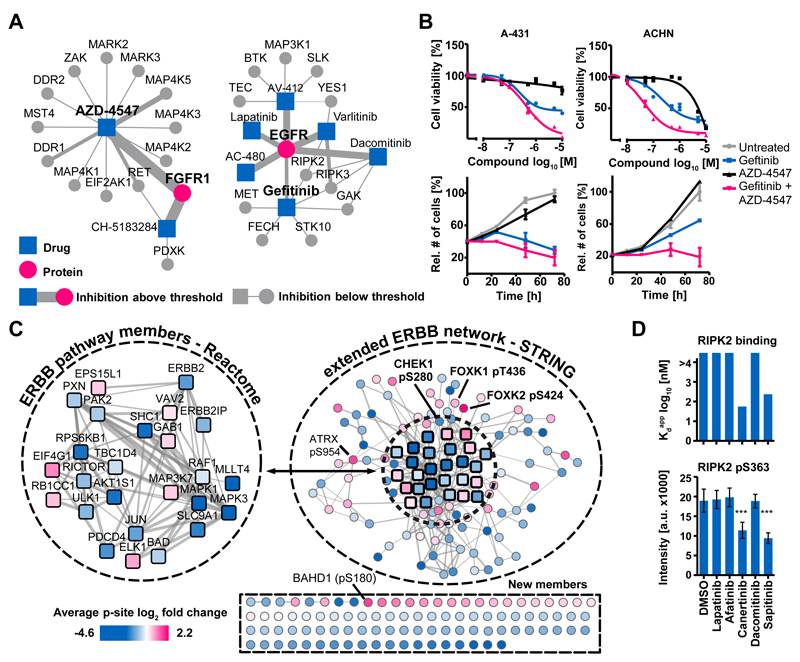
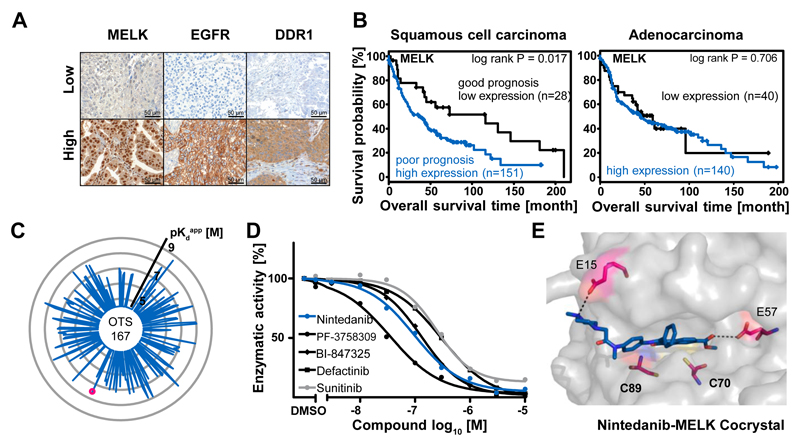
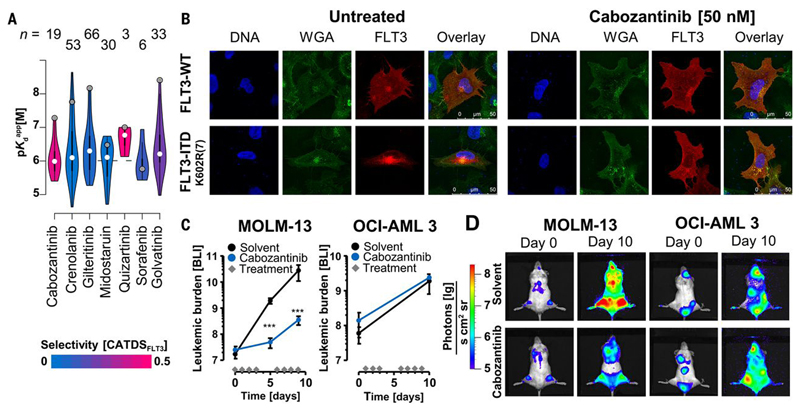
Kinase inhibitors are important cancer therapeutics. Polypharmacology is commonly observed, requiring thorough target deconvolution to understand drug mechanism of action. Using chemical proteomics, we analyzed the target spectrum of 243 clinically evaluated kinase drugs. The data revealed previously unknown targets for established drugs, offered a perspective on the "druggable" kinome, highlighted (non)kinase off-targets, and suggested potential therapeutic applications. Integration of phosphoproteomic data refined drug-affected pathways, identified response markers, and strengthened rationale for combination treatments. We exemplify translational value by discovering SIK2 (salt-inducible kinase 2) inhibitors that modulate cytokine production in primary cells, by identifying drugs against the lung cancer survival marker MELK (maternal embryonic leucine zipper kinase), and by repurposing cabozantinib to treat FLT3-ITD-positive acute myeloid leukemia. This resource, available via the ProteomicsDB database, should facilitate basic, clinical, and drug discovery research and aid clinical decision-making.
Copyright © 2017 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
New opportunities for kinase drug repurposing and target discovery.Br J Cancer. 2018 Apr;118(7):936-937. doi: 10.1038/s41416-018-0045-6. Epub 2018 Mar 16. Br J Cancer. 2018. PMID: 29545596 Free PMC article.
References
-
- Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422–439. - PubMed
-
- Karaman MW, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. - PubMed
-
- Gao Y, et al. A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem J. 2013;451:313–328. - PubMed
-
- Davis MI, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
