

What Is Our Current Understanding of PrPSc-Associated Neurotoxicity and Its Molecular Underpinnings?
- PMID: 29194372
- PMCID: PMC5750587
- DOI: 10.3390/pathogens6040063
What Is Our Current Understanding of PrPSc-Associated Neurotoxicity and Its Molecular Underpinnings?
Abstract
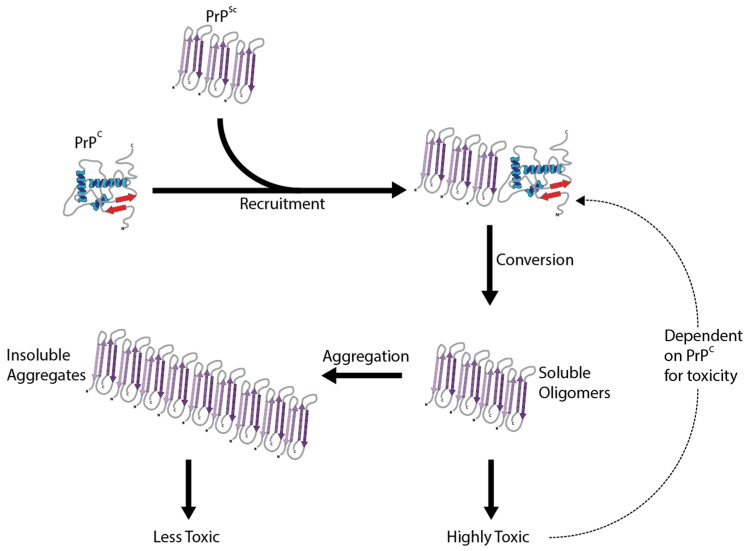
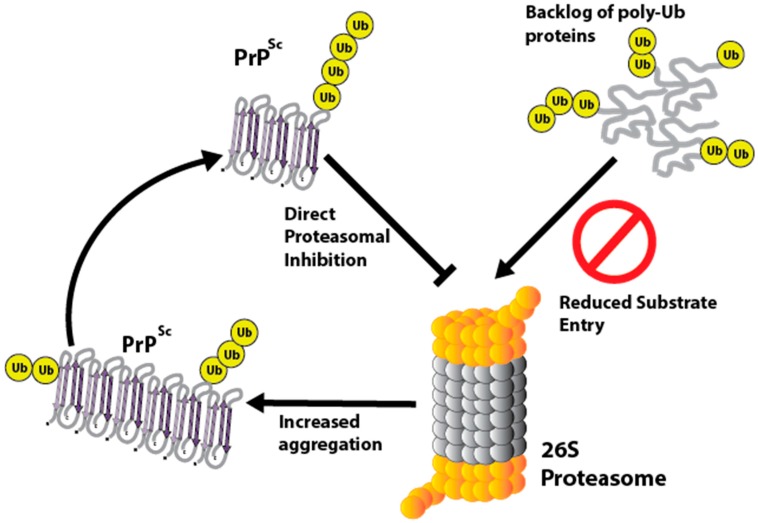
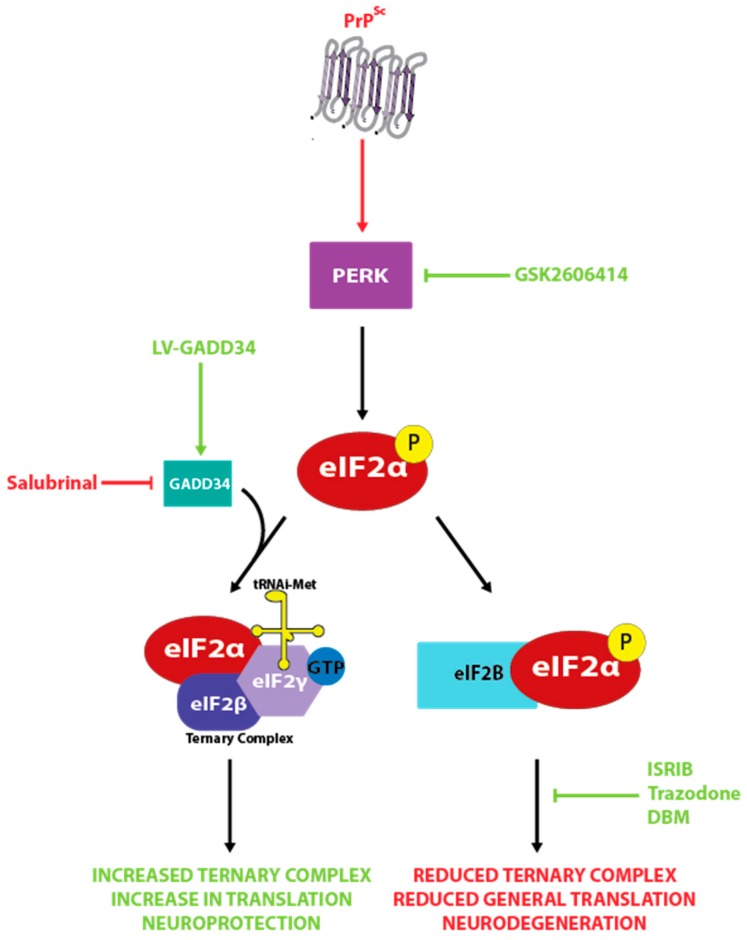
The prion diseases are a collection of fatal, transmissible neurodegenerative diseases that cause rapid onset dementia and ultimately death. Uniquely, the infectious agent is a misfolded form of the endogenous cellular prion protein, termed PrPSc. Despite the identity of the molecular agent remaining the same, PrPSc can cause a range of diseases with hereditary, spontaneous or iatrogenic aetiologies. However, the link between PrPSc and toxicity is complex, with subclinical cases of prion disease discovered, and prion neurodegeneration without obvious PrPSc deposition. The toxic mechanisms by which PrPSc causes the extensive neuropathology are still poorly understood, although recent advances are beginning to unravel the molecular underpinnings, including oxidative stress, disruption of proteostasis and induction of the unfolded protein response. This review will discuss the diseases caused by PrPSc toxicity, the nature of the toxicity of PrPSc, and our current understanding of the downstream toxic signaling events triggered by the presence of PrPSc.
Keywords: PrPSc; neurodegeneration; neurotoxicity; prion disease; proteostasis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Bamborough P., Wille H., Telling G.C., Yehiely F., Prusiner S.B., Cohen F.E. Prion protein structure and scrapie replication: Theoretical, spectroscopic, and genetic investigations. Cold Spring Harb. Symp. Quant. Biol. 1996;61:495–509. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
