

p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1
- PMID: 29196462
- PMCID: PMC5748971
- DOI: 10.1083/jcb.201701049
p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1
Abstract
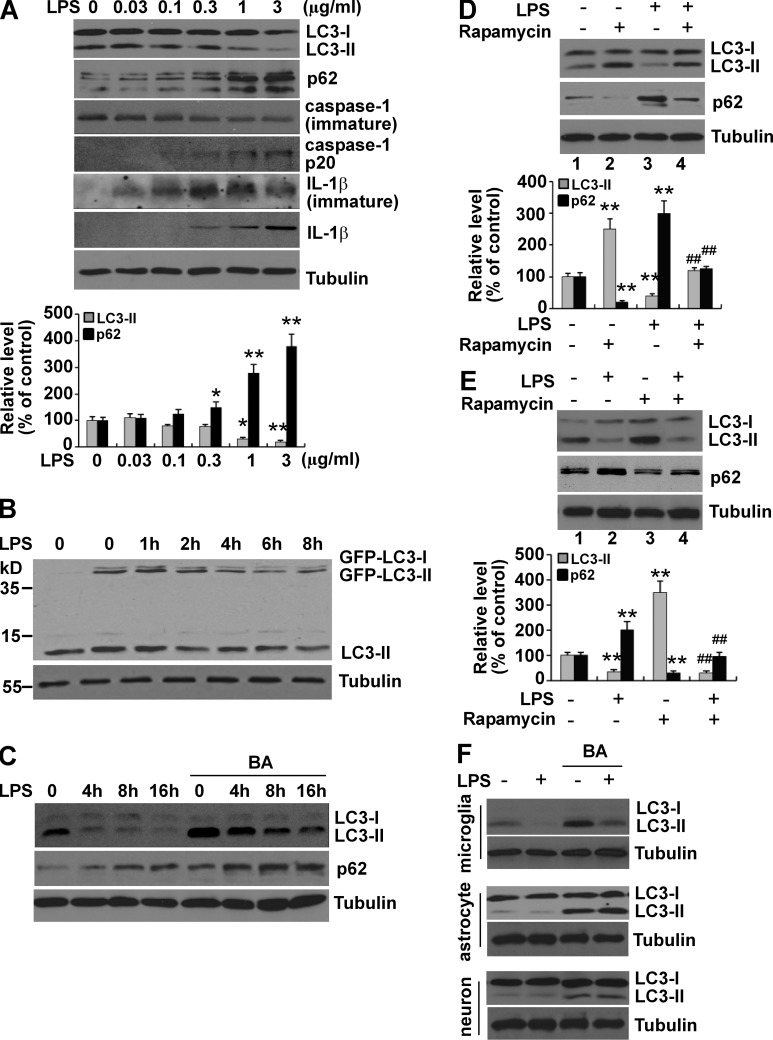
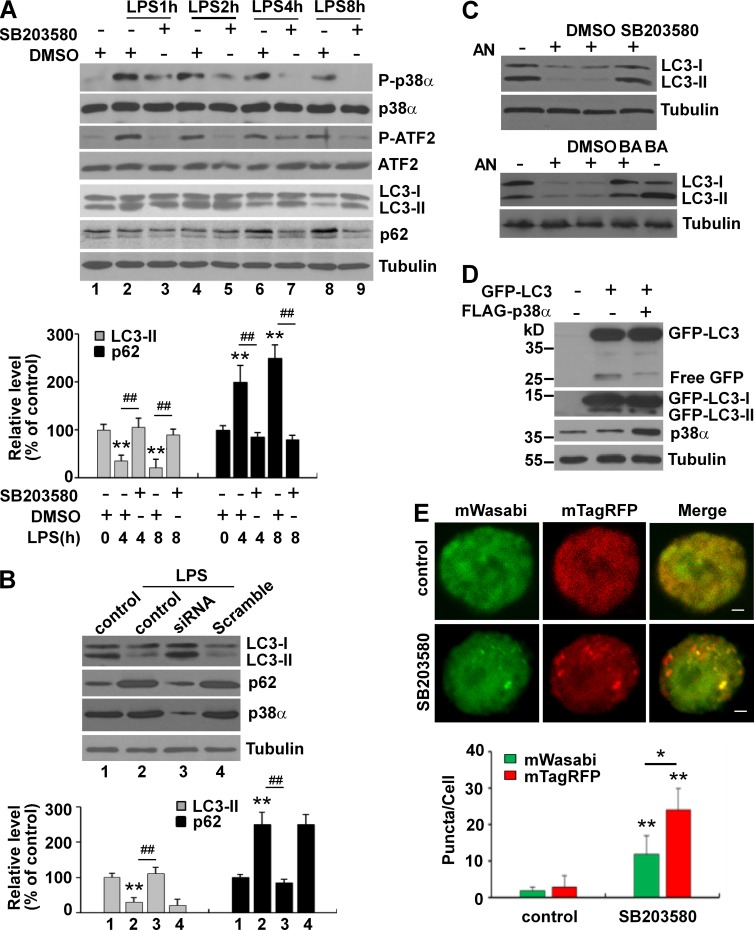
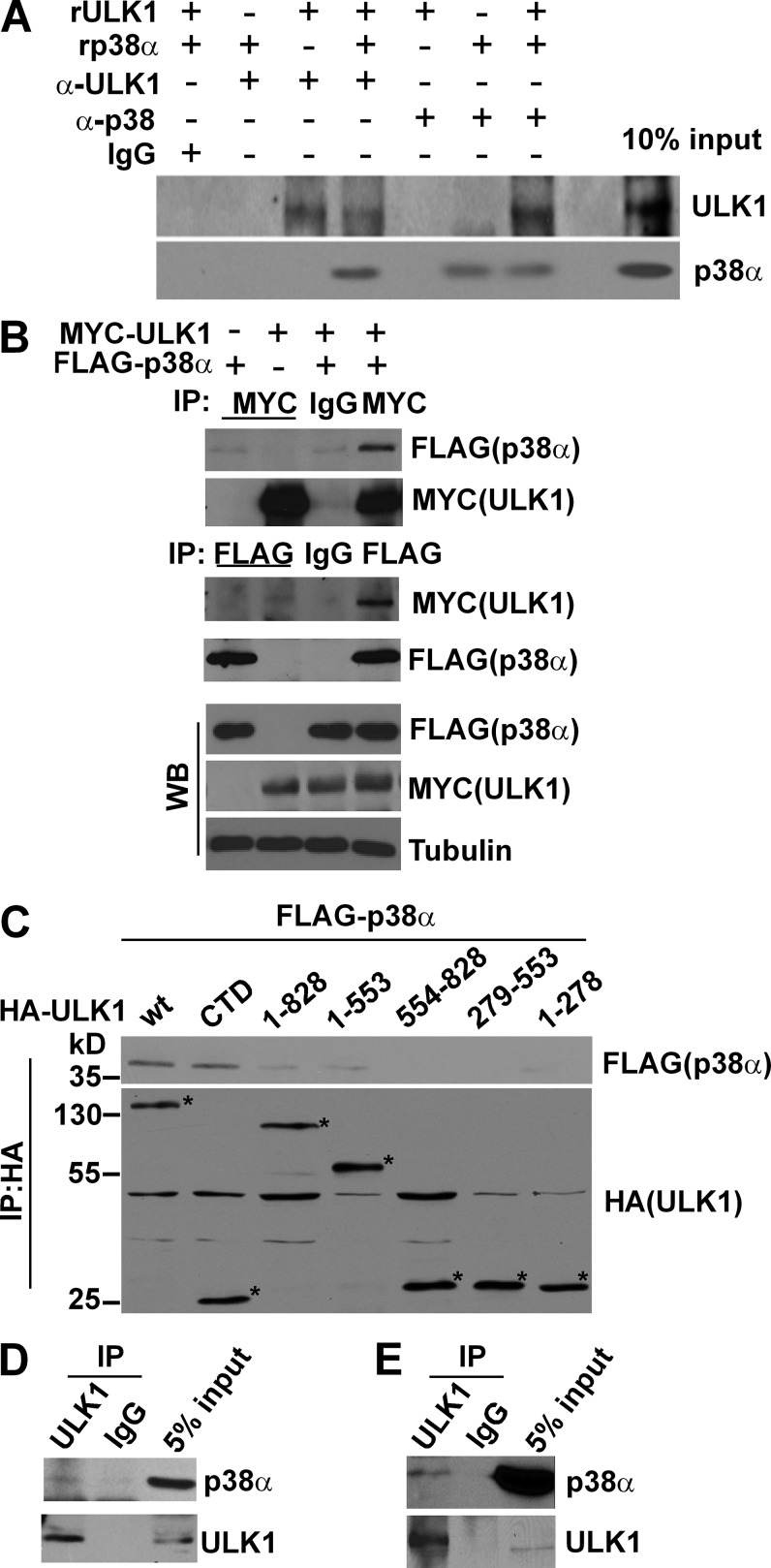
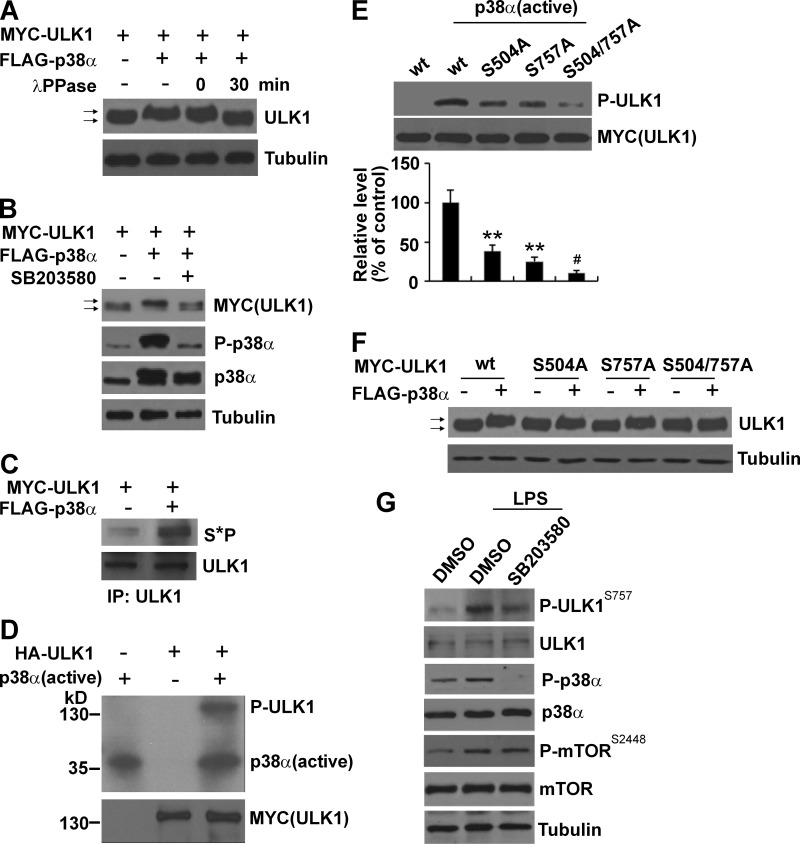
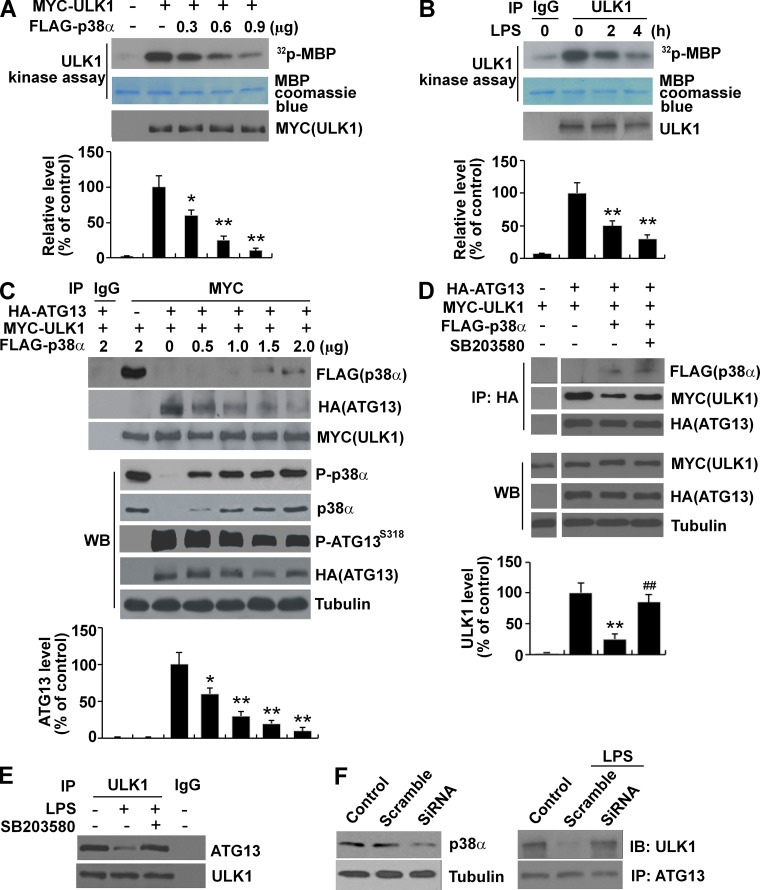
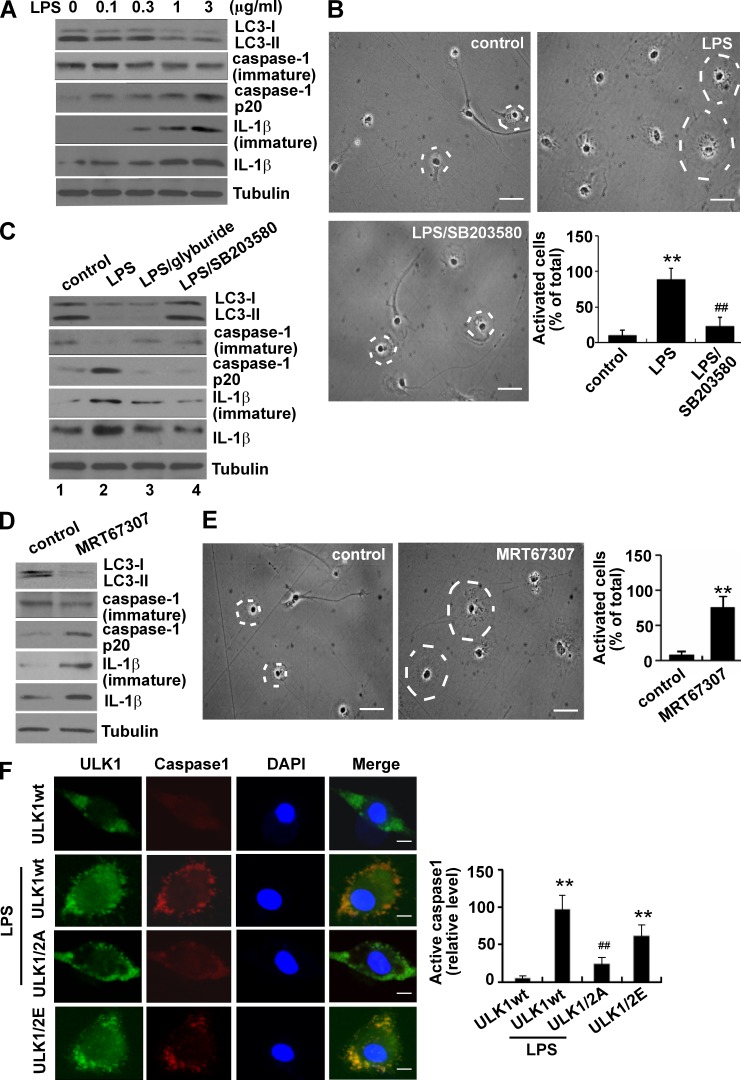
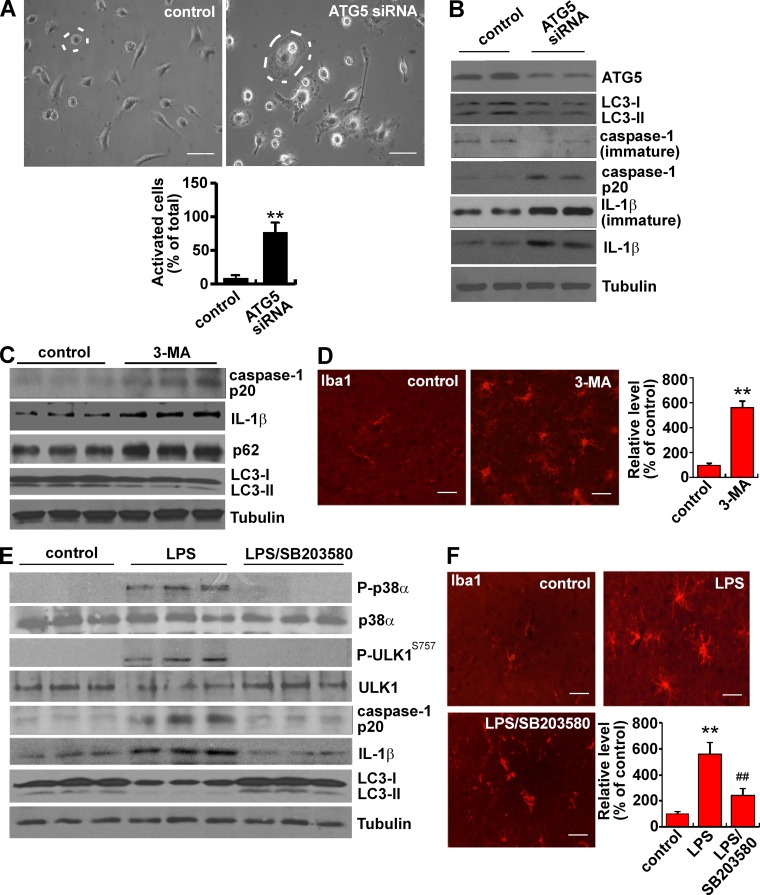
Inflammation and autophagy are two critical cellular processes. The relationship between these two processes is complex and includes the suppression of inflammation by autophagy. However, the signaling mechanisms that relieve this autophagy-mediated inhibition of inflammation to permit a beneficial inflammatory response remain unknown. We find that LPS triggers p38α mitogen-activated protein kinase (MAPK)-dependent phosphorylation of ULK1 in microglial cells. This phosphorylation inhibited ULK1 kinase activity, preventing it from binding to the downstream effector ATG13, and reduced autophagy in microglia. Consistently, p38α MAPK activity is required for LPS-induced morphological changes and the production of IL-1β by primary microglia in vitro and in the brain, which correlates with the p38α MAPK-dependent inhibition of autophagy. Furthermore, inhibition of ULK1 alone was sufficient to promote an inflammatory response in the absence of any overt inflammatory stimulation. Thus, our study reveals a molecular mechanism that enables the initial TLR4-triggered signaling pathway to inhibit autophagy and optimize inflammatory responses, providing new understanding into the mechanistic basis of the neuroinflammatory process.
© 2018 He et al.
Figures
Comment in
-
Release the autophage brake on inflammation: The MAPK14/p38α-ULK1 pedal.Autophagy. 2018;14(6):1097-1098. doi: 10.1080/15548627.2018.1446626. Epub 2018 May 11. Autophagy. 2018. PMID: 29749797 Free PMC article.
References
-
- Bachstetter A.D., Xing B., de Almeida L., Dimayuga E.R., Watterson D.M., and Van Eldik L.J.. 2011. Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Aβ). J. Neuroinflammation. 8:79 10.1186/1742-2094-8-79 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
