

Microfluidic isoform sequencing shows widespread splicing coordination in the human transcriptome
- PMID: 29196558
- PMCID: PMC5793787
- DOI: 10.1101/gr.230516.117
Microfluidic isoform sequencing shows widespread splicing coordination in the human transcriptome
Abstract
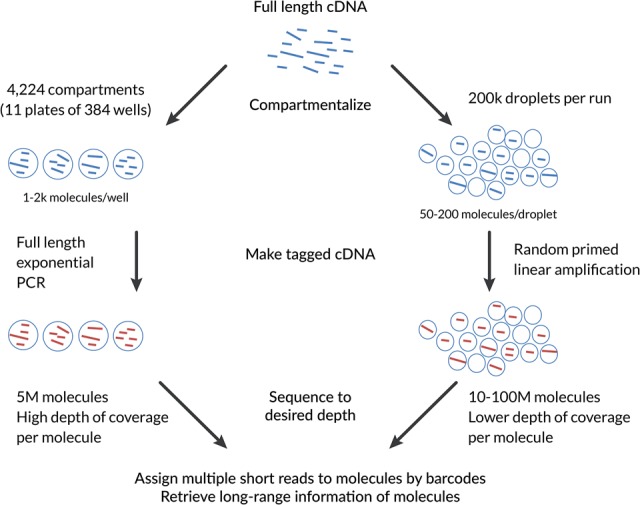
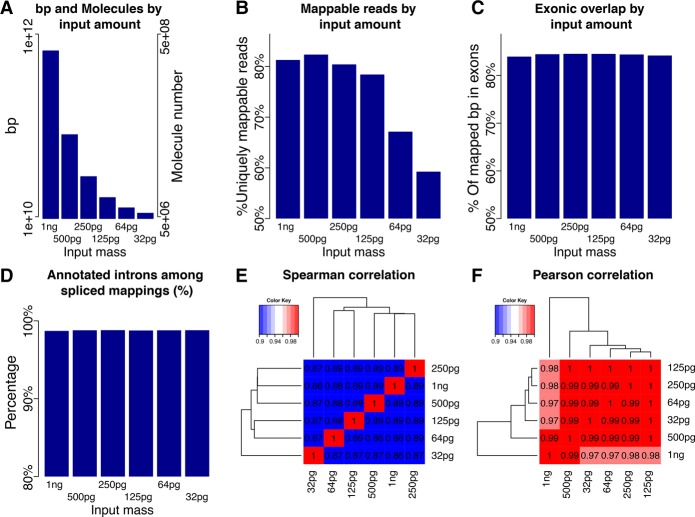
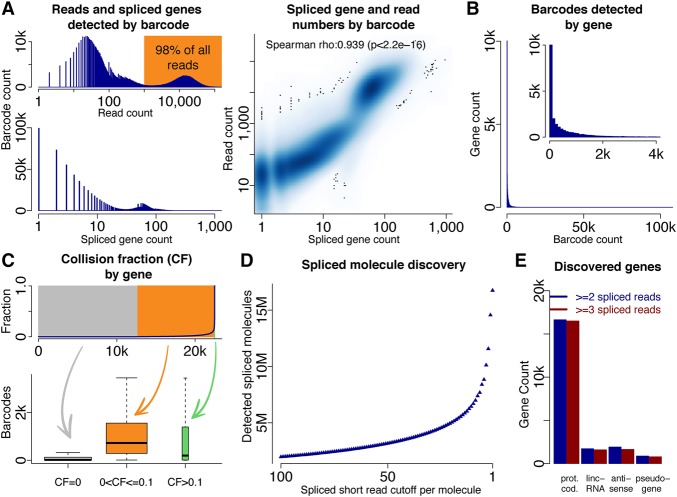
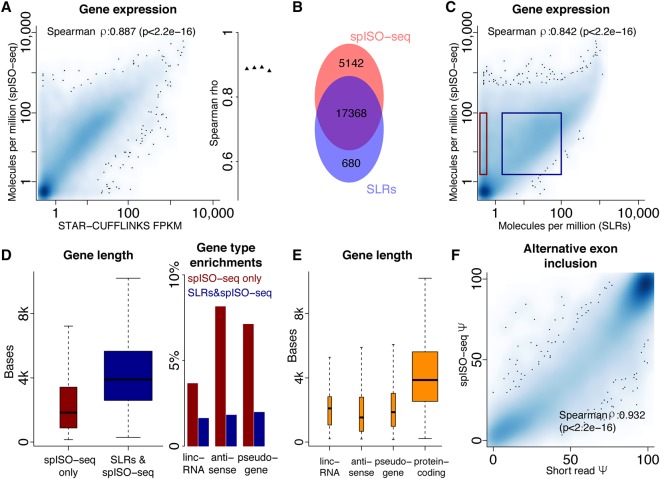
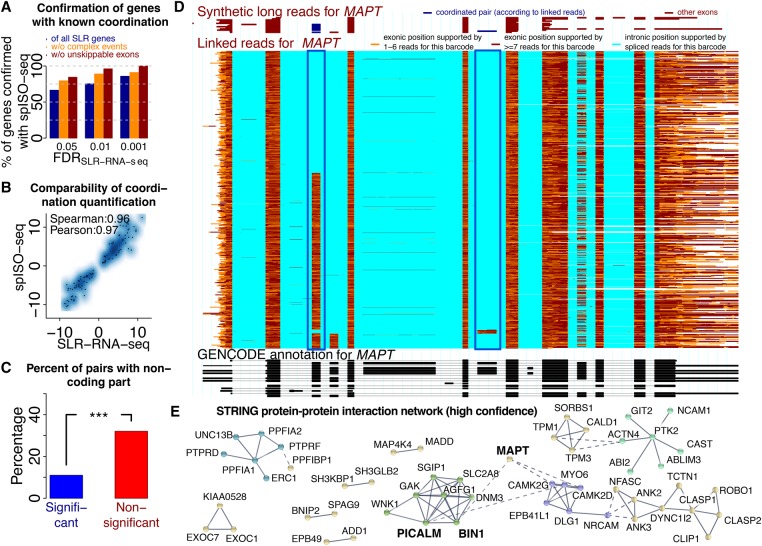
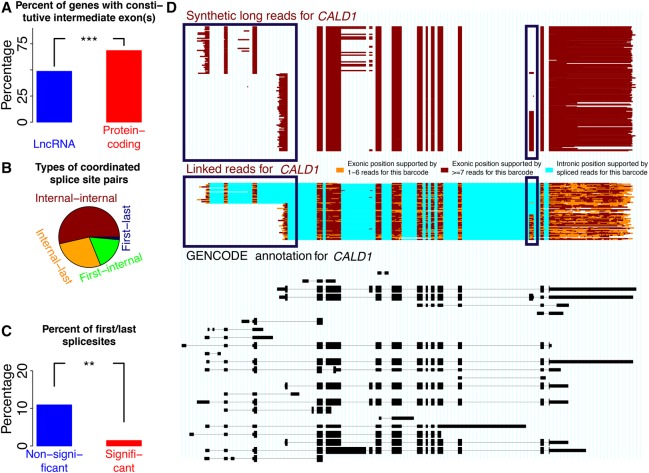
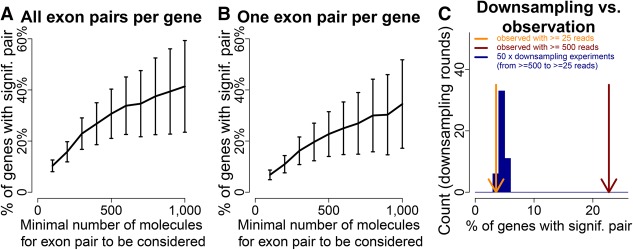
Understanding transcriptome complexity is crucial for understanding human biology and disease. Technologies such as Synthetic long-read RNA sequencing (SLR-RNA-seq) delivered 5 million isoforms and allowed assessing splicing coordination. Pacific Biosciences and Oxford Nanopore increase throughput also but require high input amounts or amplification. Our new droplet-based method, sparse isoform sequencing (spISO-seq), sequences 100k-200k partitions of 10-200 molecules at a time, enabling analysis of 10-100 million RNA molecules. SpISO-seq requires less than 1 ng of input cDNA, limiting or removing the need for prior amplification with its associated biases. Adjusting the number of reads devoted to each molecule reduces sequencing lanes and cost, with little loss in detection power. The increased number of molecules expands our understanding of isoform complexity. In addition to confirming our previously published cases of splicing coordination (e.g., BIN1), the greater depth reveals many new cases, such as MAPT Coordination of internal exons is found to be extensive among protein coding genes: 23.5%-59.3% (95% confidence interval) of highly expressed genes with distant alternative exons exhibit coordination, showcasing the need for long-read transcriptomics. However, coordination is less frequent for noncoding sequences, suggesting a larger role of splicing coordination in shaping proteins. Groups of genes with coordination are involved in protein-protein interactions with each other, raising the possibility that coordination facilitates complex formation and/or function. We also find new splicing coordination types, involving initial and terminal exons. Our results provide a more comprehensive understanding of the human transcriptome and a general, cost-effective method to analyze it.
© 2018 Tilgner et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
BRIE: transcriptome-wide splicing quantification in single cells.Genome Biol. 2017 Jun 27;18(1):123. doi: 10.1186/s13059-017-1248-5. Genome Biol. 2017. PMID: 28655331 Free PMC article.
-
Transcript Profiling Using Long-Read Sequencing Technologies.Methods Mol Biol. 2018;1783:121-147. doi: 10.1007/978-1-4939-7834-2_6. Methods Mol Biol. 2018. PMID: 29767360
-
Reconstruction of the full-length transcriptome atlas using PacBio Iso-Seq provides insight into the alternative splicing in Gossypium australe.BMC Plant Biol. 2019 Aug 19;19(1):365. doi: 10.1186/s12870-019-1968-7. BMC Plant Biol. 2019. PMID: 31426739 Free PMC article.
-
Decoding co-/post-transcriptional complexities of plant transcriptomes and epitranscriptome using next-generation sequencing technologies.Biochem Soc Trans. 2020 Dec 18;48(6):2399-2414. doi: 10.1042/BST20190492. Biochem Soc Trans. 2020. PMID: 33196096 Review.
-
Identification of Splice Variants and Isoforms in Transcriptomics and Proteomics.Annu Rev Biomed Data Sci. 2023 Aug 10;6:357-376. doi: 10.1146/annurev-biodatasci-020722-044021. Annu Rev Biomed Data Sci. 2023. PMID: 37561601 Free PMC article. Review.
Cited by
-
Getting the Entire Message: Progress in Isoform Sequencing.Front Genet. 2019 Aug 16;10:709. doi: 10.3389/fgene.2019.00709. eCollection 2019. Front Genet. 2019. PMID: 31475029 Free PMC article. Review.
-
acorde unravels functionally interpretable networks of isoform co-usage from single cell data.Nat Commun. 2022 Apr 5;13(1):1828. doi: 10.1038/s41467-022-29497-w. Nat Commun. 2022. PMID: 35383181 Free PMC article.
-
Repeat-associated RNA structure and aberrant splicing.Biochim Biophys Acta Gene Regul Mech. 2019 Nov-Dec;1862(11-12):194405. doi: 10.1016/j.bbagrm.2019.07.006. Epub 2019 Jul 16. Biochim Biophys Acta Gene Regul Mech. 2019. PMID: 31323433 Free PMC article. Review.
-
Targeted DNA-seq and RNA-seq of Reference Samples with Short-read and Long-read Sequencing.Sci Data. 2024 Aug 16;11(1):892. doi: 10.1038/s41597-024-03741-y. Sci Data. 2024. PMID: 39152166 Free PMC article.
-
Targeted transcriptome analysis using synthetic long read sequencing uncovers isoform reprograming in the progression of colon cancer.Commun Biol. 2021 Apr 27;4(1):506. doi: 10.1038/s42003-021-02024-1. Commun Biol. 2021. PMID: 33907296 Free PMC article.
References
-
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 57: 289–300.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources