

Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy
- PMID: 29196579
- PMCID: PMC5754647
- DOI: 10.1212/WNL.0000000000004762
Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy
Abstract
Objective: To characterize the phenotypic spectrum, molecular genetic findings, and functional consequences of pathogenic variants in early-onset KCNT1 epilepsy.
Methods: We identified a cohort of 31 patients with epilepsy of infancy with migrating focal seizures (EIMFS) and screened for variants in KCNT1 using direct Sanger sequencing, a multiple-gene next-generation sequencing panel, and whole-exome sequencing. Additional patients with non-EIMFS early-onset epilepsy in whom we identified KCNT1 variants on local diagnostic multiple gene panel testing were also included. When possible, we performed homology modeling to predict the putative effects of variants on protein structure and function. We undertook electrophysiologic assessment of mutant KCNT1 channels in a xenopus oocyte model system.
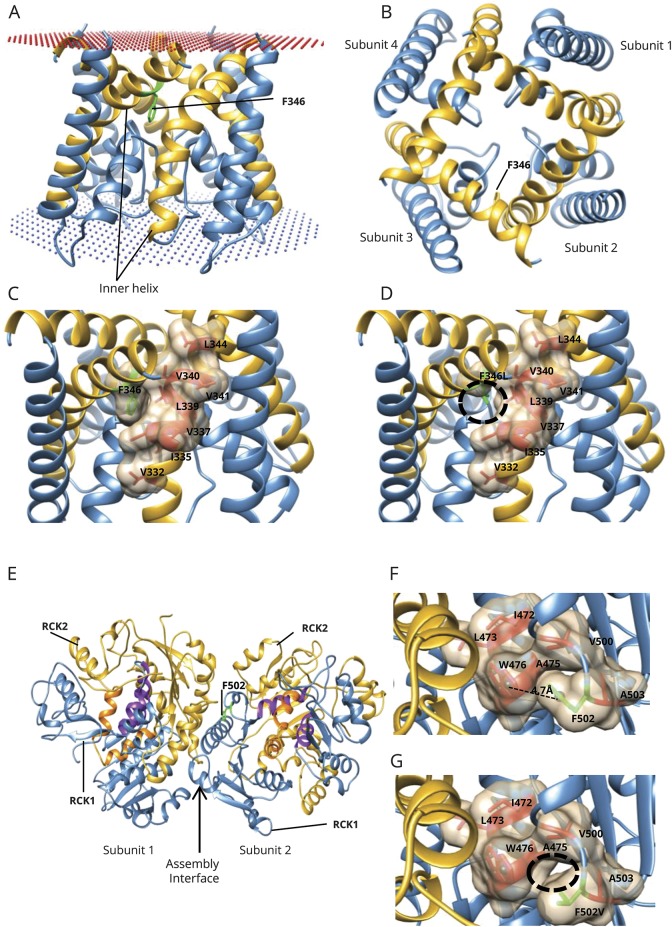
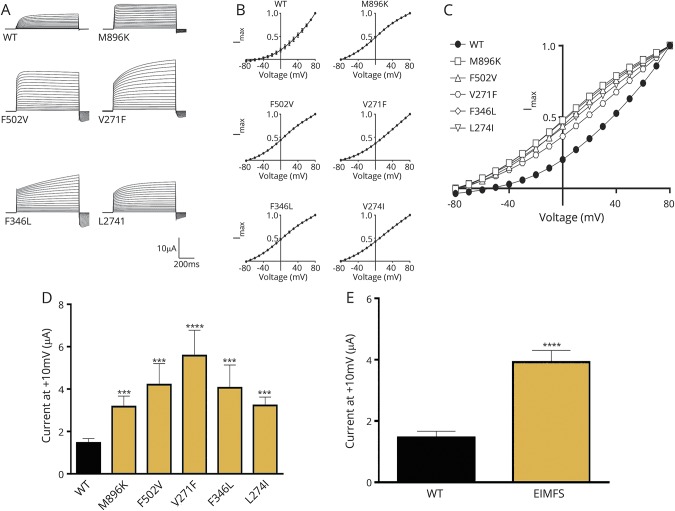
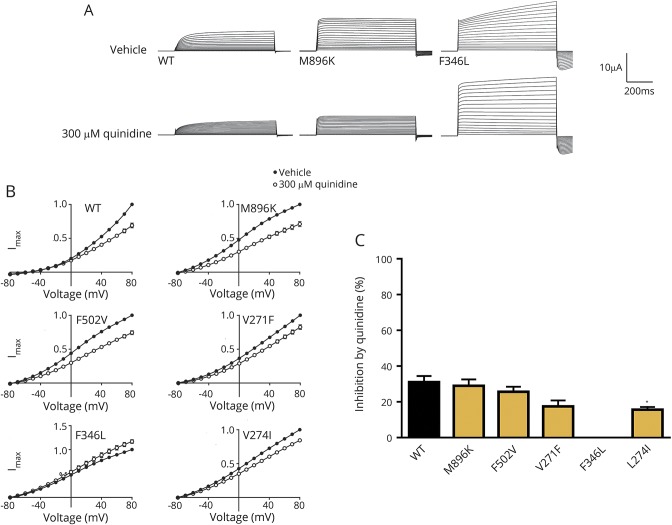
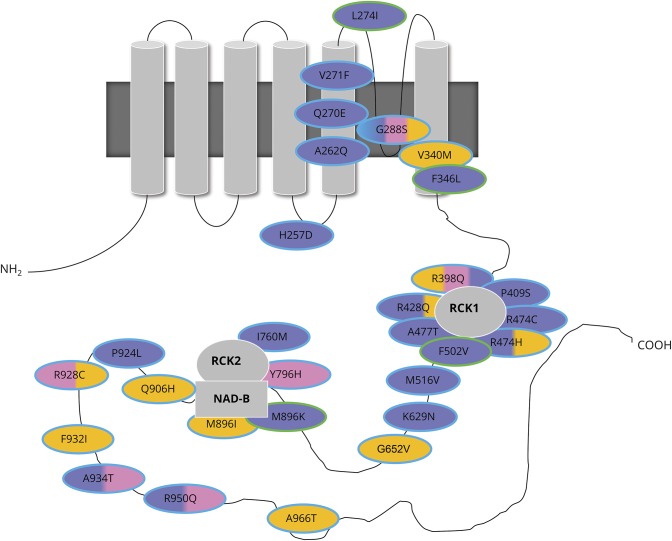
Results: We identified pathogenic variants in KCNT1 in 12 patients, 4 of which are novel. Most variants occurred de novo. Ten patients had a clinical diagnosis of EIMFS, and the other 2 presented with early-onset severe nocturnal frontal lobe seizures. Three patients had a trial of quinidine with good clinical response in 1 patient. Computational modeling analysis implicates abnormal pore function (F346L) and impaired tetramer formation (F502V) as putative disease mechanisms. All evaluated KCNT1 variants resulted in marked gain of function with significantly increased channel amplitude and variable blockade by quinidine.
Conclusions: Gain-of-function KCNT1 pathogenic variants cause a spectrum of severe focal epilepsies with onset in early infancy. Currently, genotype-phenotype correlations are unclear, although clinical outcome is poor for the majority of cases. Further elucidation of disease mechanisms may facilitate the development of targeted treatments, much needed for this pharmacoresistant genetic epilepsy.
Copyright © 2017 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures
References
-
- Fukuoka M, Kuki I, Kawawaki H, et al. Quinidine therapy for West syndrome with KCNT1 mutation: a case report. Brain Dev 2017;39:80–83. - PubMed
-
- Ishii A, Shioda M, Okumura A, et al. A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy. Gene 2013;531:467–471. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources