

Using the genome aggregation database, computational pathogenicity prediction tools, and patch clamp heterologous expression studies to demote previously published long QT syndrome type 1 mutations from pathogenic to benign
- PMID: 29197658
- PMCID: PMC6383800
- DOI: 10.1016/j.hrthm.2017.11.032
Using the genome aggregation database, computational pathogenicity prediction tools, and patch clamp heterologous expression studies to demote previously published long QT syndrome type 1 mutations from pathogenic to benign
Abstract
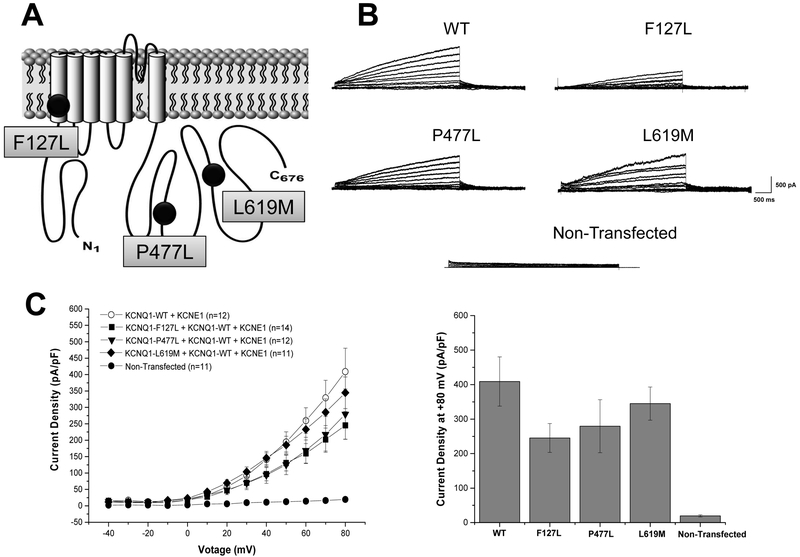
Background: Mutations in the KCNQ1-encoded Kv7.1 potassium channel cause long QT syndrome (LQTS) type 1 (LQT1). It has been suggested that ∼10%-20% of rare LQTS case-derived variants in the literature may have been published erroneously as LQT1-causative mutations and may be "false positives."
Objective: The purpose of this study was to determine which previously published KCNQ1 case variants are likely false positives.
Methods: A list of all published, case-derived KCNQ1 missense variants (MVs) was compiled. The occurrence of each MV within the Genome Aggregation Database (gnomAD) was assessed. Eight in silico tools were used to predict each variant's pathogenicity. Case-derived variants that were either (1) too frequently found in gnomAD or (2) absent in gnomAD but predicted to be pathogenic by ≤2 tools were considered potential false positives. Three of these variants were characterized functionally using whole-cell patch clamp technique.
Results: Overall, there were 244 KCNQ1 case-derived MVs. Of these, 29 (12%) were seen in ≥10 individuals in gnomAD and are demotable. However, 157 of 244 MVs (64%) were absent in gnomAD. Of these, 7 (4%) were predicted to be pathogenic by ≤2 tools, 3 of which we characterized functionally. There was no significant difference in current density between heterozygous KCNQ1-F127L, -P477L, or -L619M variant-containing channels compared to KCNQ1-WT.
Conclusion: This study offers preliminary evidence for the demotion of 32 (13%) previously published LQT1 MVs. Of these, 29 were demoted because of their frequent sighting in gnomAD. Additionally, in silico analysis and in vitro functional studies have facilitated the demotion of 3 ultra-rare MVs (F127L, P477L, L619M).
Keywords: Arrhythmia; Genetics; Heart arrest; KCNQ1; Long QT syndrome; Pediatrics.
Copyright © 2017 Heart Rhythm Society. Published by Elsevier Inc. All rights reserved.
Figures

Comment in
-
The phenotype is equally important in promoting variants from benign to pathogenic as well as in demoting variants from pathogenic to benign.Heart Rhythm. 2018 Apr;15(4):562-563. doi: 10.1016/j.hrthm.2018.01.007. Epub 2018 Jan 6. Heart Rhythm. 2018. PMID: 29317317 No abstract available.
References
-
- Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circulation Research 2015;116:1919–1936. - PubMed
-
- Rook MB, Alshinawi CB, Groenewegen WA, Van Gelder IC, Van Ginneken AC, Jongsma HJ, Mannens MM, Wilde AA. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovascular Research 1999;44:507–517. - PubMed
-
- Nakajima-Taniguchi C, Matsui H, Fujio Y, Nagata S, Kishimoto T, Yamauchi-Takihara K. Novel missense mutation in Cardiac Troponin T gene found in Japanese patient with hypertrophic cardiomyopathy. Journal of Molecular and Cellular Cardiology 1997;29:839–843. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
