

Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer
- PMID: 29198524
- PMCID: PMC5878932
- DOI: 10.1016/j.cell.2017.10.044
Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer
Abstract
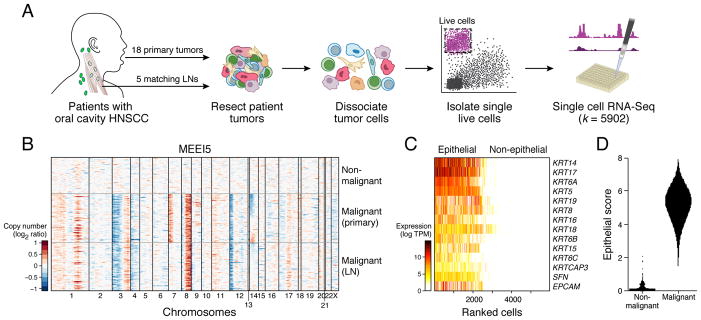
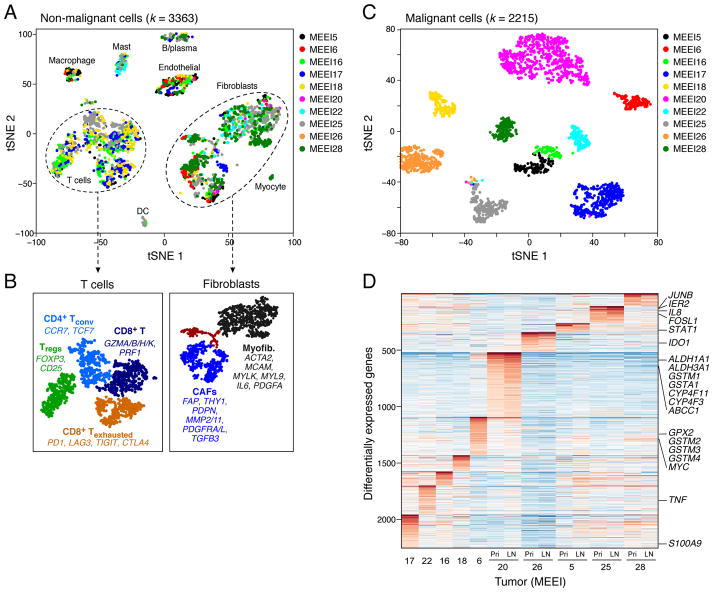
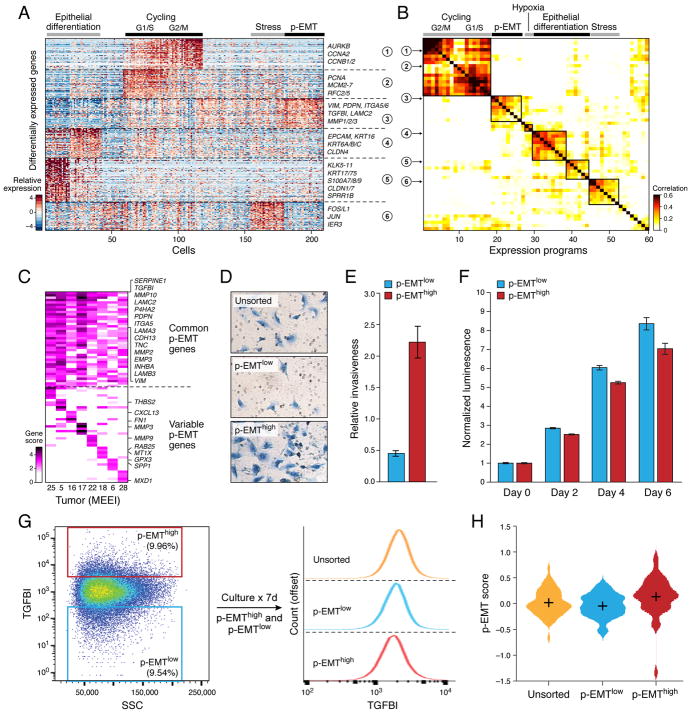
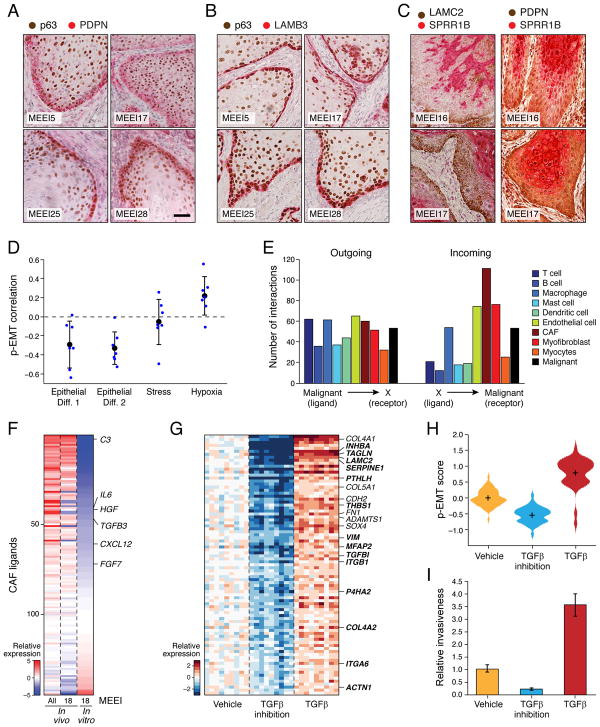
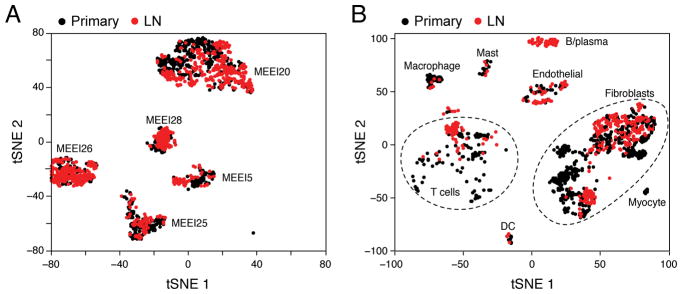
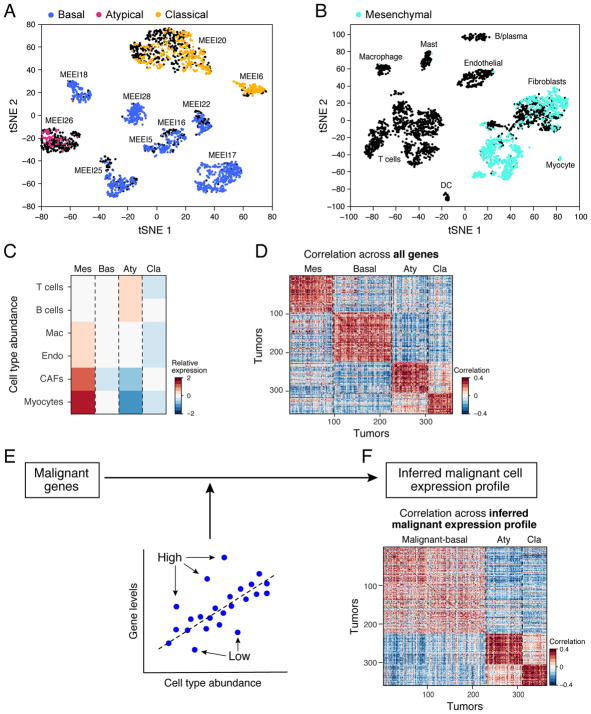
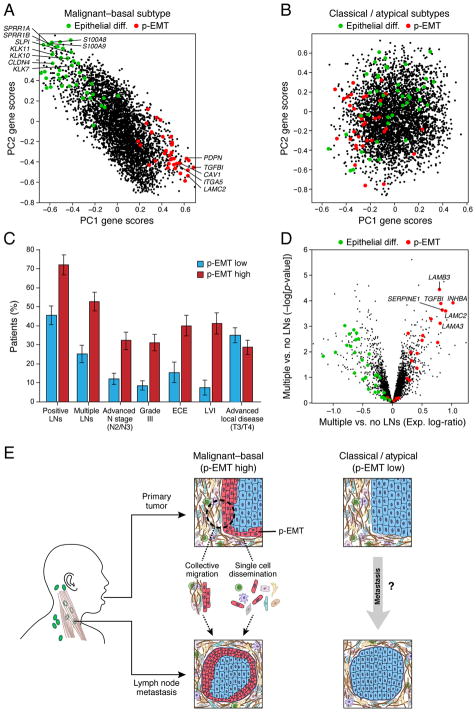
The diverse malignant, stromal, and immune cells in tumors affect growth, metastasis, and response to therapy. We profiled transcriptomes of ∼6,000 single cells from 18 head and neck squamous cell carcinoma (HNSCC) patients, including five matched pairs of primary tumors and lymph node metastases. Stromal and immune cells had consistent expression programs across patients. Conversely, malignant cells varied within and between tumors in their expression of signatures related to cell cycle, stress, hypoxia, epithelial differentiation, and partial epithelial-to-mesenchymal transition (p-EMT). Cells expressing the p-EMT program spatially localized to the leading edge of primary tumors. By integrating single-cell transcriptomes with bulk expression profiles for hundreds of tumors, we refined HNSCC subtypes by their malignant and stromal composition and established p-EMT as an independent predictor of nodal metastasis, grade, and adverse pathologic features. Our results provide insight into the HNSCC ecosystem and define stromal interactions and a p-EMT program associated with metastasis.
Keywords: epithelial-to-mesenchymal transition; head and neck squamous cell carcinoma; intra-tumoral heterogeneity; metastasis; scRNA-seq; single-cell RNA sequencing; tumor microenvironment.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
Comment in
-
When it comes to genomic analysis of tumours, don't buy in bulk.Br J Cancer. 2018 May;118(10):1281-1282. doi: 10.1038/s41416-018-0096-8. Epub 2018 May 10. Br J Cancer. 2018. PMID: 29743553 Free PMC article.
-
Nasopharyngeal carcinoma joins the single-cell party.Cancer Commun (Lond). 2020 Sep;40(9):453-455. doi: 10.1002/cac2.12091. Epub 2020 Aug 25. Cancer Commun (Lond). 2020. PMID: 32841535 Free PMC article. No abstract available.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
