

Mutations in TUBB4B Cause a Distinctive Sensorineural Disease
- PMID: 29198720
- PMCID: PMC5812887
- DOI: 10.1016/j.ajhg.2017.10.010
Mutations in TUBB4B Cause a Distinctive Sensorineural Disease
Abstract
Leber congenital amaurosis (LCA) is a neurodegenerative disease of photoreceptor cells that causes blindness within the first year of life. It occasionally occurs in syndromic metabolic diseases and plurisystemic ciliopathies. Using exome sequencing in a multiplex family and three simplex case subjects with an atypical association of LCA with early-onset hearing loss, we identified two heterozygous mutations affecting Arg391 in β-tubulin 4B isotype-encoding (TUBB4B). Inspection of the atomic structure of the microtubule (MT) protofilament reveals that the β-tubulin Arg391 residue contributes to a binding pocket that interacts with α-tubulin contained in the longitudinally adjacent αβ-heterodimer, consistent with a role in maintaining MT stability. Functional analysis in cultured cells overexpressing FLAG-tagged wild-type or mutant TUBB4B as well as in primary skin-derived fibroblasts showed that the mutant TUBB4B is able to fold, form αβ-heterodimers, and co-assemble into the endogenous MT lattice. However, the dynamics of growing MTs were consistently altered, showing that the mutations have a significant dampening impact on normal MT growth. Our findings provide a link between sensorineural disease and anomalies in MT behavior and describe a syndromic LCA unrelated to ciliary dysfunction.
Keywords: Leber congenital amaurosis; TUBB4B; abnormal dynamics of microtubule growth; de novo mutations; dominant mutations; early-onset sensorineural hearing loss; mosaicism; retino-cochlear tubulinopathy.
Copyright © 2017 American Society of Human Genetics. All rights reserved.
Figures
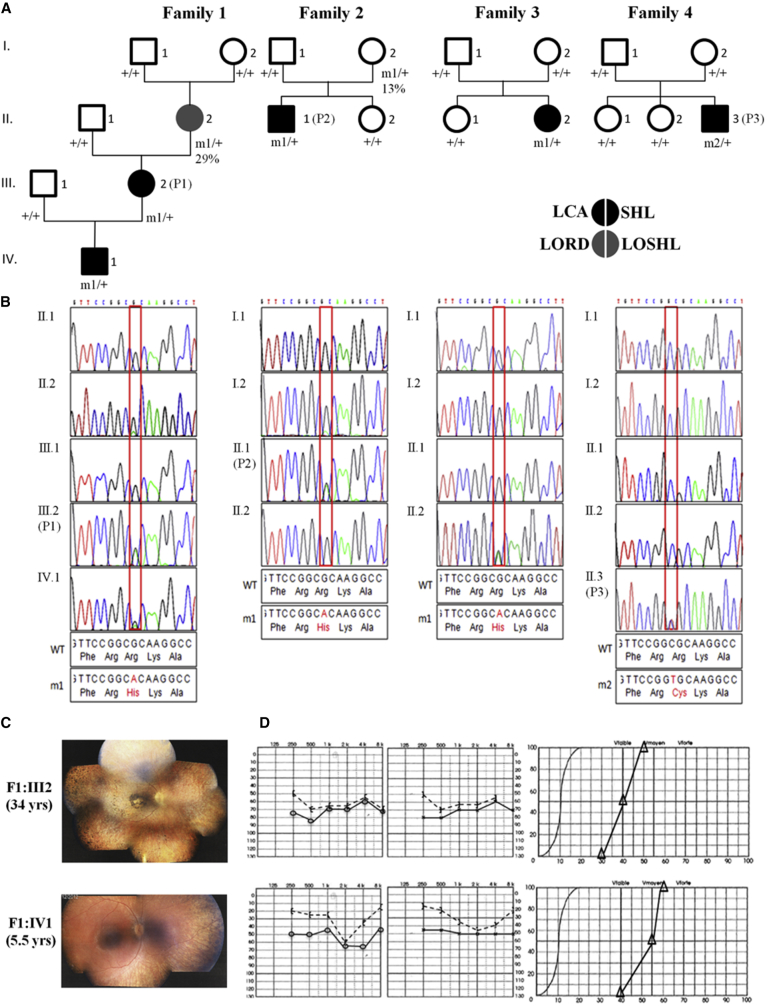
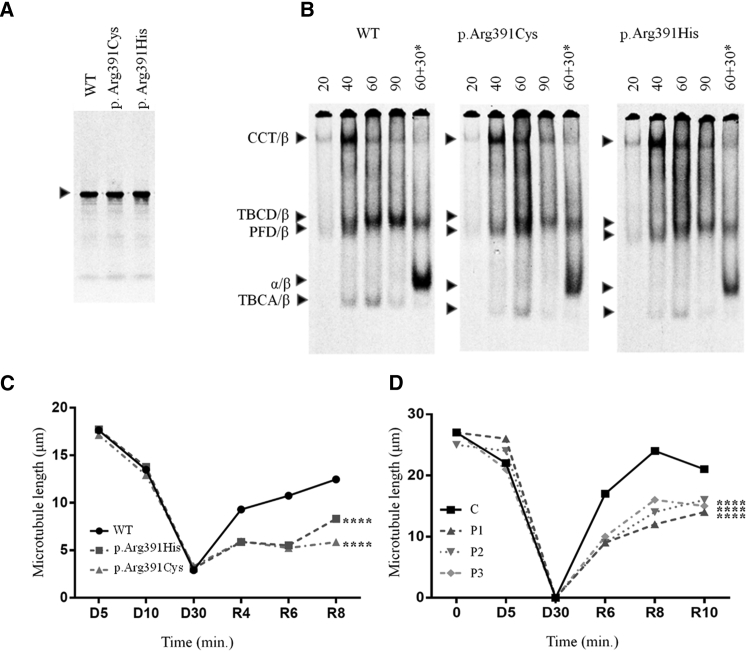
References
-
- Gadadhar S., Bodakuntla S., Natarajan K., Janke C. The tubulin code at a glance. J. Cell Sci. 2017;130:1347–1353. - PubMed
-
- Leber T. Ueber Retinitis pigmentosa und angeborene Amaurose. Archiv fur Opthalmologie. 1869;15:1–25.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
