

Effects of exercise on obesity-induced mitochondrial dysfunction in skeletal muscle
- PMID: 29200899
- PMCID: PMC5709473
- DOI: 10.4196/kjpp.2017.21.6.567
Effects of exercise on obesity-induced mitochondrial dysfunction in skeletal muscle
Abstract
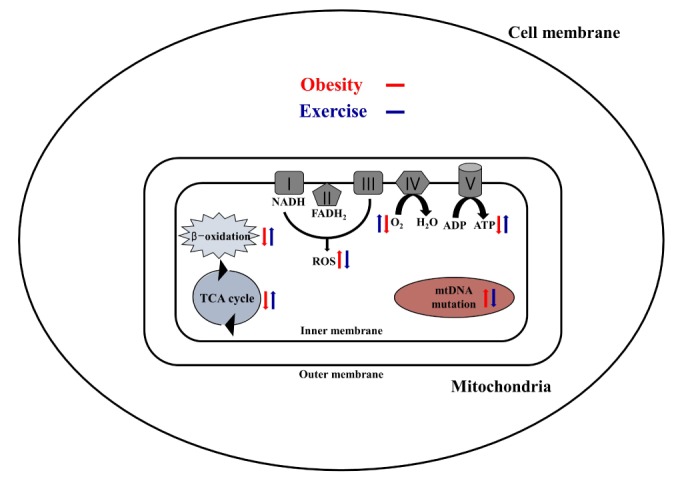
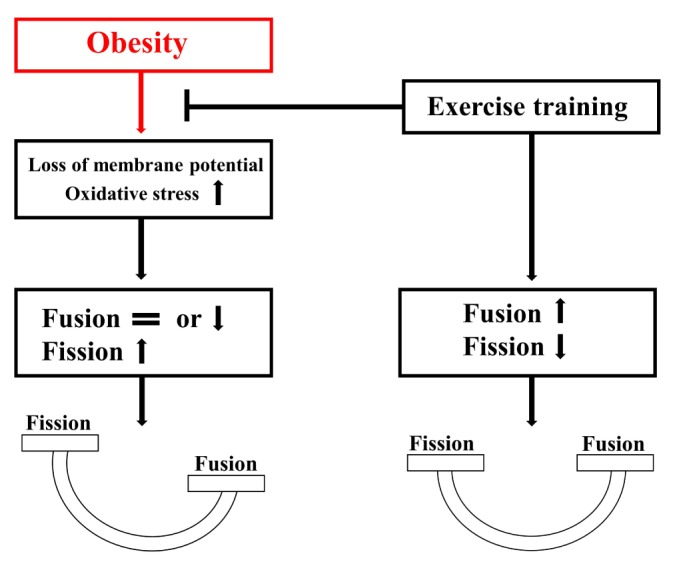
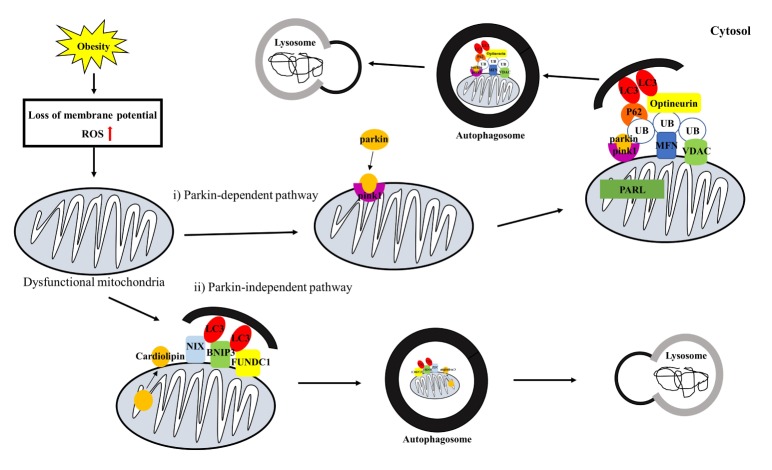
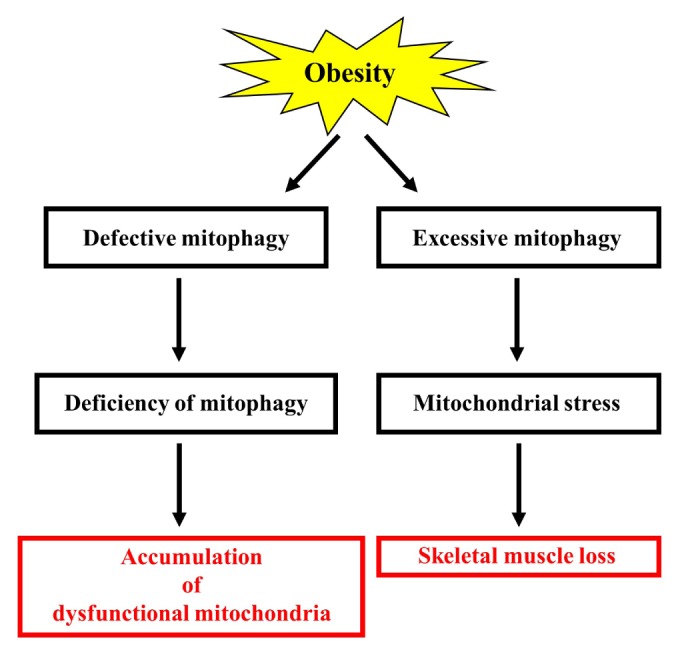
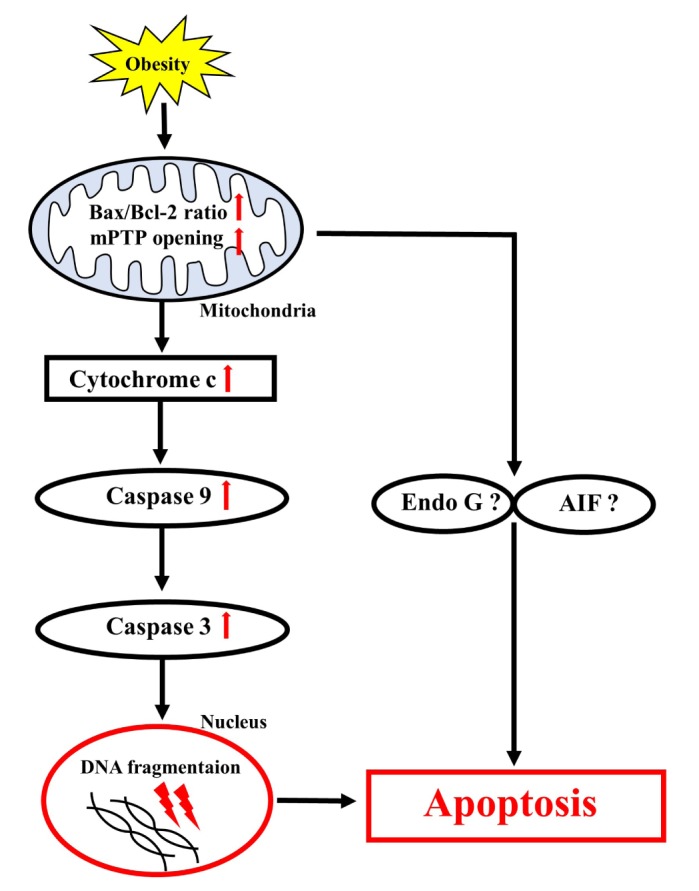
Obesity is known to induce inhibition of glucose uptake, reduction of lipid metabolism, and progressive loss of skeletal muscle function, which are all associated with mitochondrial dysfunction in skeletal muscle. Mitochondria are dynamic organelles that regulate cellular metabolism and bioenergetics, including ATP production via oxidative phosphorylation. Due to these critical roles of mitochondria, mitochondrial dysfunction results in various diseases such as obesity and type 2 diabetes. Obesity is associated with impairment of mitochondrial function (e.g., decrease in O2 respiration and increase in oxidative stress) in skeletal muscle. The balance between mitochondrial fusion and fission is critical to maintain mitochondrial homeostasis in skeletal muscle. Obesity impairs mitochondrial dynamics, leading to an unbalance between fusion and fission by favorably shifting fission or reducing fusion proteins. Mitophagy is the catabolic process of damaged or unnecessary mitochondria. Obesity reduces mitochondrial biogenesis in skeletal muscle and increases accumulation of dysfunctional cellular organelles, suggesting that mitophagy does not work properly in obesity. Mitochondrial dysfunction and oxidative stress are reported to trigger apoptosis, and mitochondrial apoptosis is induced by obesity in skeletal muscle. It is well known that exercise is the most effective intervention to protect against obesity. Although the cellular and molecular mechanisms by which exercise protects against obesity-induced mitochondrial dysfunction in skeletal muscle are not clearly elucidated, exercise training attenuates mitochondrial dysfunction, allows mitochondria to maintain the balance between mitochondrial dynamics and mitophagy, and reduces apoptotic signaling in obese skeletal muscle.
Keywords: Exercise; Mitochondria; Obesity; Skeletal Muscle.
Conflict of interest statement
CONFLICTS OF INTEREST: The authors declare no conflicts of interest.
Figures
References
-
- Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, Abraham JP, Abu-Rmeileh NM, Achoki T, AlBuhairan FS, Alemu ZA, Alfonso R, Ali MK, Ali R, Guzman NA, Ammar W, Anwari P, Banerjee A, Barquera S, Basu S, Bennett DA, Bhutta Z, Blore J, Cabral N, Nonato IC, Chang JC, Chowdhury R, Courville KJ, Criqui MH, Cundiff DK, Dabhadkar KC, Dandona L, Davis A, Dayama A, Dharmaratne SD, Ding EL, Durrani AM, Esteghamati A, Farzadfar F, Fay DF, Feigin VL, Flaxman A, Forouzanfar MH, Goto A, Green MA, Gupta R, Hafezi-Nejad N, Hankey GJ, Harewood HC, Havmoeller R, Hay S, Hernandez L, Husseini A, Idrisov BT, Ikeda N, Islami F, Jahangir E, Jassal SK, Jee SH, Jeffreys M, Jonas JB, Kabagambe EK, Khalifa SE, Kengne AP, Khader YS, Khang YH, Kim D, Kimokoti RW, Kinge JM, Kokubo Y, Kosen S, Kwan G, Lai T, Leinsalu M, Li Y, Liang X, Liu S, Logroscino G, Lotufo PA, Lu Y, Ma J, Mainoo NK, Mensah GA, Merriman TR, Mokdad AH, Moschandreas J, Naghavi M, Naheed A, Nand D, Narayan KM, Nelson EL, Neuhouser ML, Nisar MI, Ohkubo T, Oti SO, Pedroza A, Prabhakaran D, Roy N, Sampson U, Seo H, Sepanlou SG, Shibuya K, Shiri R, Shiue I, Singh GM, Singh JA, Skirbekk V, Stapelberg NJ, Sturua L, Sykes BL, Tobias M, Tran BX, Trasande L, Toyoshima H, van de Vijver S, Vasankari TJ, Veerman JL, Velasquez-Melendez G, Vlassov VV, Vollset SE, Vos T, Wang C, Wang X, Weiderpass E, Werdecker A, Wright JL, Yang YC, Yatsuya H, Yoon J, Yoon SJ, Zhao Y, Zhou M, Zhu S, Lopez AD, Murray CJ, Gakidou E. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:766–781. - PMC - PubMed
-
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. - PubMed
-
- Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. - PMC - PubMed
-
- Greene NP, Nilsson MI, Washington TA, Lee DE, Brown LA, Papineau AM, Shimkus KL, Greene ES, Crouse SF, Fluckey JD. Impaired exercise-induced mitochondrial biogenesis in the obese Zucker rat, despite PGC-1α induction, is due to compromised mitochondrial translation elongation. Am J Physiol Endocrinol Metab. 2014;306:E503–E511. - PubMed
-
- Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacín M, Zorzano A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14:1405–1415. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
