

qPCR primer design revisited
- PMID: 29201647
- PMCID: PMC5702850
- DOI: 10.1016/j.bdq.2017.11.001
qPCR primer design revisited
Abstract
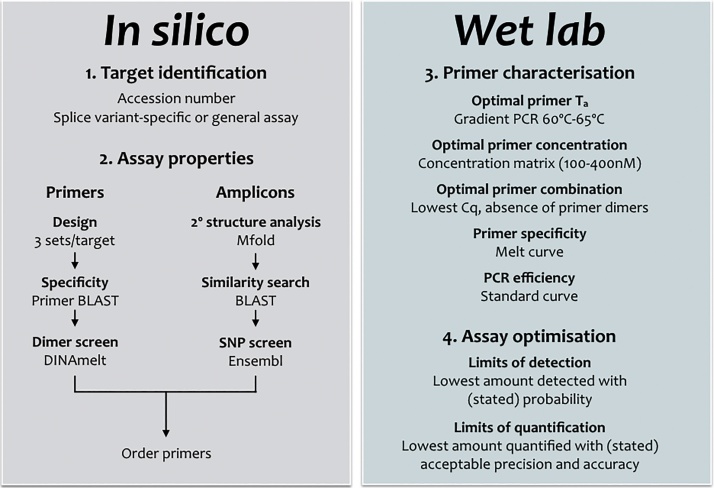
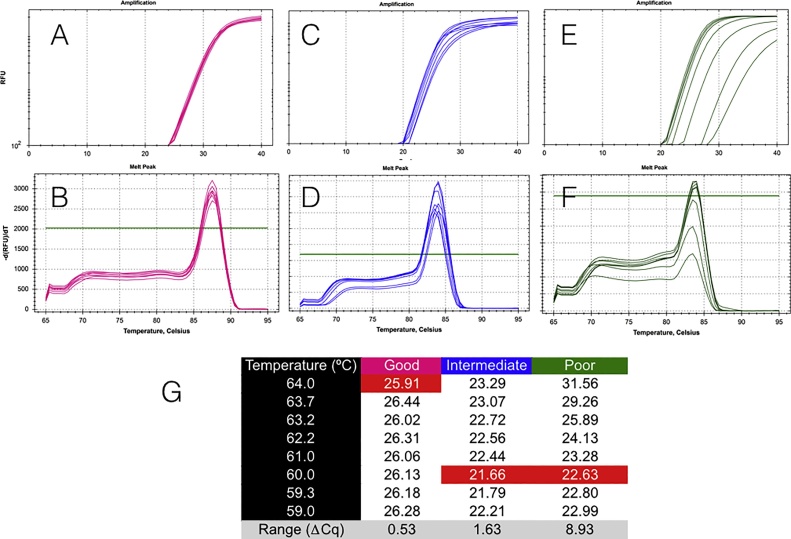
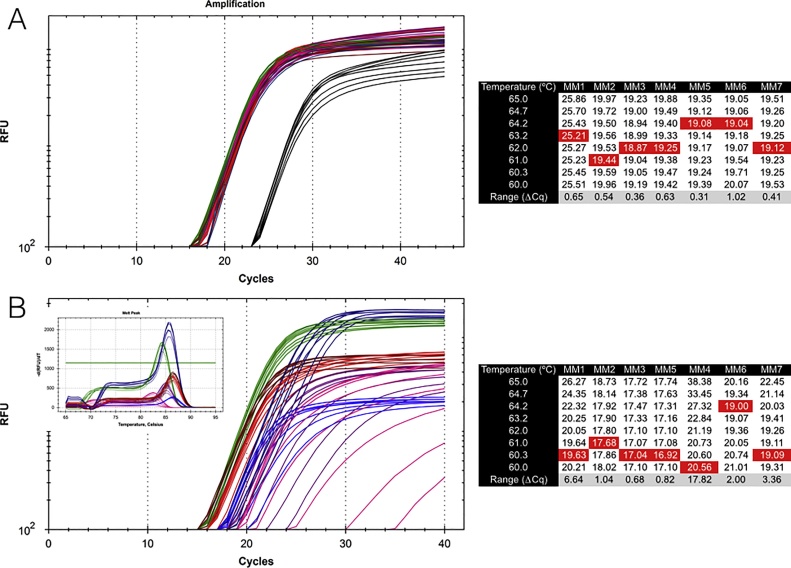
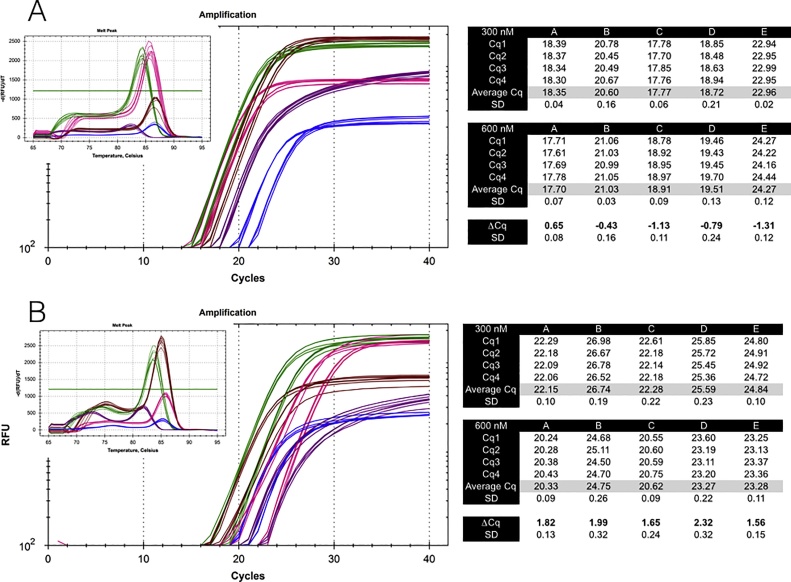
Primers are arguably the single most critical components of any PCR assay, as their properties control the exquisite specificity and sensitivity that make this method uniquely powerful. Consequently, poor design combined with failure to optimise reaction conditions is likely to result in reduced technical precision and false positive or negative detection of amplification targets. Despite the framework provided by the MIQE guidelines and the accessibility of wide-ranging support from peer-reviewed publications, books and online sources as well as commercial companies, the design of many published assays continues to be less than optimal: primers often lack intended specificity, can form dimers, compete with template secondary structures at the primer binding sites or hybridise only within a narrow temperature range. We present an overview of the main steps in the primer design workflow, with data that illustrate some of the unexpected variability that often occurs when theory is translated into practice. We also strongly urge researchers to report as much information about their assays as possible in their publications.
Keywords: Assay design; MIQE; Oligonucleotides; Real-time PCR.
Figures
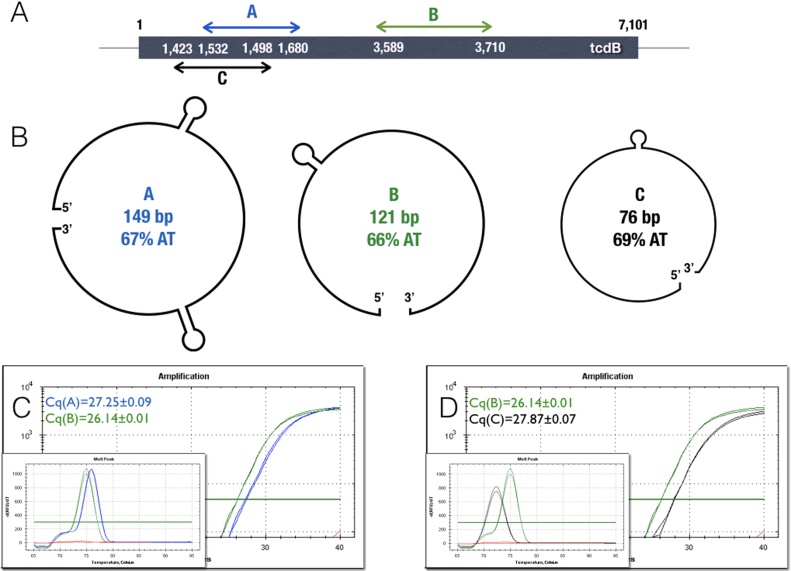
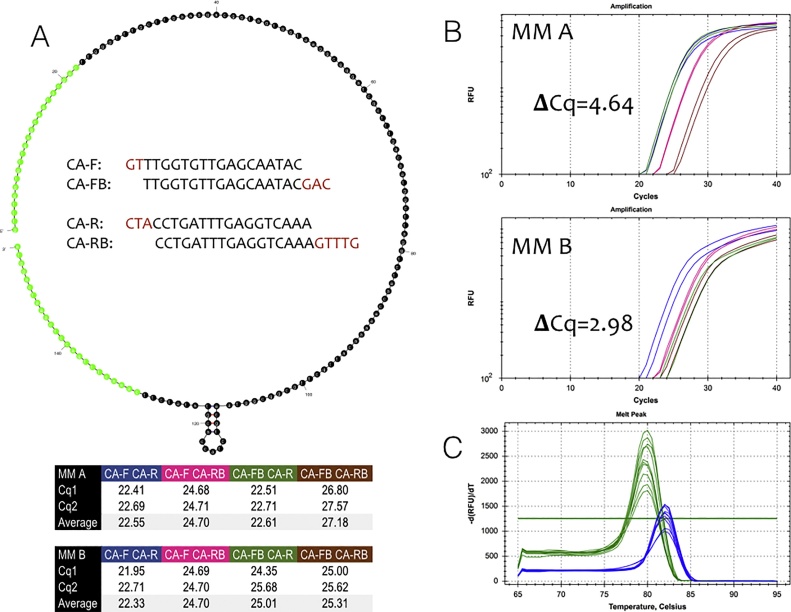
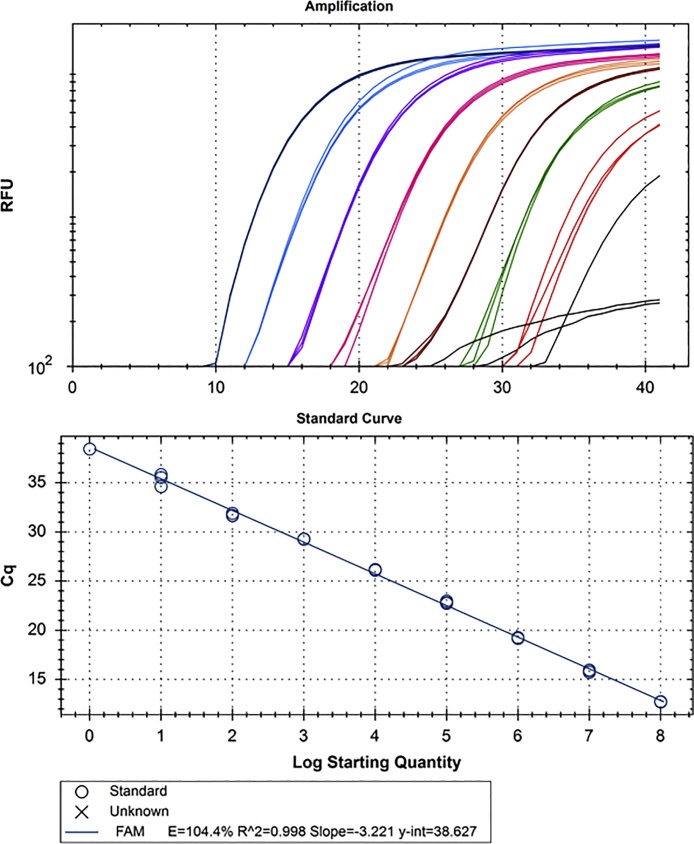
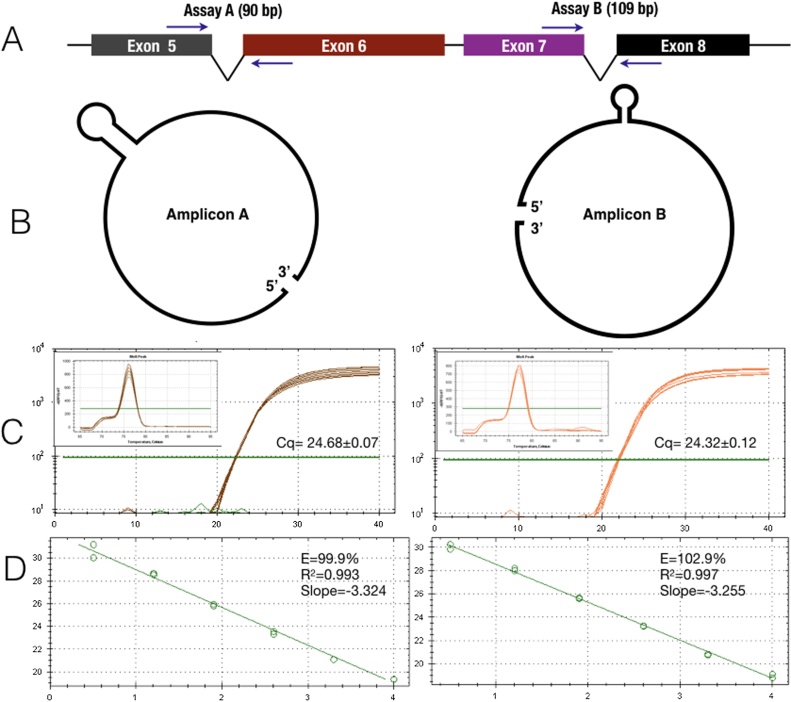
References
-
- Raggi C.C., Verderio P., Pazzagli M., Marubini E., Simi L., Pinzani P. An Italian program of external quality control for quantitative assays based on real-time PCR with Taq-Man probes. Clin. Chem. Lab. Med. 2005;43:542–548. - PubMed
-
- Buzard G.S., Baker D., Wolcott M.J., Norwood D.A., Dauphin L.A. Multi-platform comparison of ten commercial master mixes for probe-based real-time polymerase chain reaction detection of bioterrorism threat agents for surge preparedness. Forensic Sci. Int. 2012;223:292–297. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources