

Emergence and Spread of Epidemic Multidrug-Resistant Pseudomonas aeruginosa
- PMID: 29202180
- PMCID: PMC5726472
- DOI: 10.1093/gbe/evx243
Emergence and Spread of Epidemic Multidrug-Resistant Pseudomonas aeruginosa
Abstract
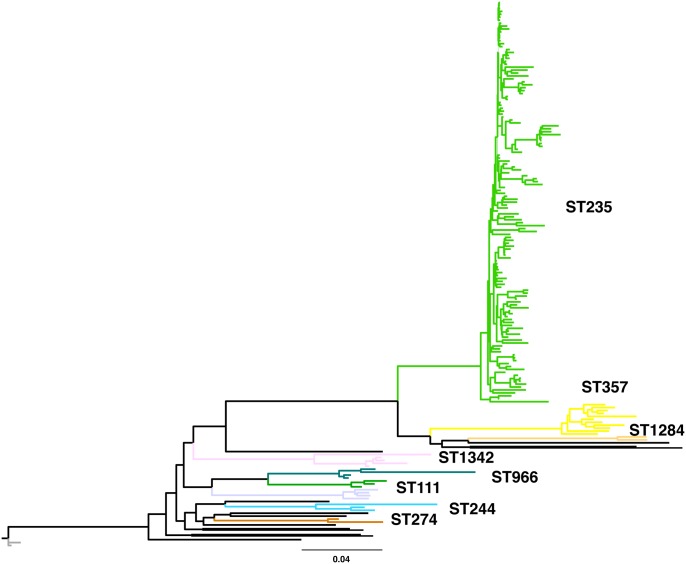
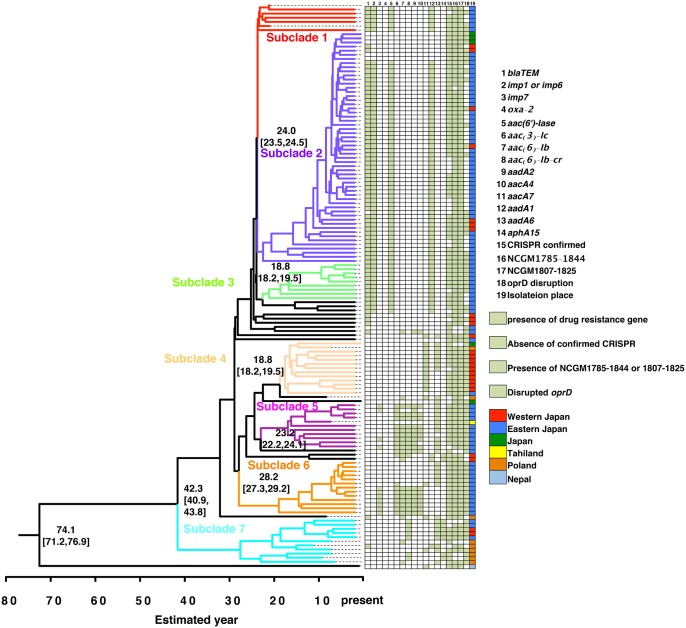
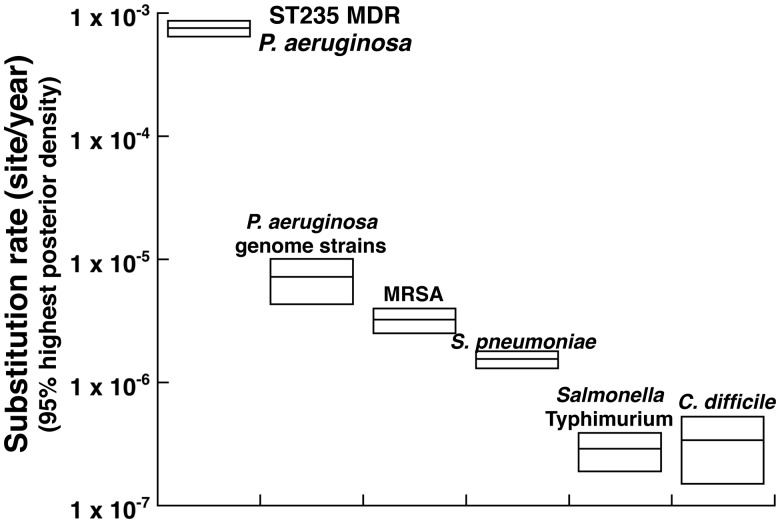
Pseudomonas aeruginosa (P. aeruginosa) is one of the most common nosocomial pathogens worldwide. Although the emergence of multidrug-resistant (MDR) P. aeruginosa is a critical problem in medical practice, the key features involved in the emergence and spread of MDR P. aeruginosa remain unknown. This study utilized whole genome sequence (WGS) analyses to define the population structure of 185 P. aeruginosa clinical isolates from several countries. Of these 185 isolates, 136 were categorized into sequence type (ST) 235, one of the most common types worldwide. Phylogenetic analysis showed that these isolates fell within seven subclades. Each subclade harbors characteristic drug resistance genes and a characteristic genetic background confined to a geographic location, suggesting that clonal expansion following antibiotic exposure is the driving force in generating the population structure of MDR P. aeruginosa. WGS analyses also showed that the substitution rate was markedly higher in ST235 MDR P. aeruginosa than in other strains. Notably, almost all ST235 isolates harbor the specific type IV secretion system and very few or none harbor the CRISPR/CAS system. These findings may help explain the mechanism underlying the emergence and spread of ST235 P. aeruginosa as the predominant MDR lineage.
Keywords: Pseudomonas aeruginosa; multidrug-resistance; population structure; whole genome sequence.
© The Author(s) 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410. - PubMed
-
- Alvarez-Martinez CE, Christie PJ.. 2009. Biological diversity of prokaryotic type IV secretion systems. Microbiol Mol Biol Rev. 73(4):775–808.http://dx.doi.org/10.1128/MMBR.00023-09 - DOI - PMC - PubMed
-
- Bhaya D, Davison M, Barrangou R.. 2011. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 45:273–297.http://dx.doi.org/10.1146/annurev-genet-110410-132430 - DOI - PubMed
-
- Chen FJ, Lo HJ.. 2003. Molecular mechanisms of fluoroquinolone resistance. J Microbiol Immunol Infect. 36(1):1–9. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
