

Hyposialylated IgG activates endothelial IgG receptor FcγRIIB to promote obesity-induced insulin resistance
- PMID: 29202472
- PMCID: PMC5749535
- DOI: 10.1172/JCI89333
Hyposialylated IgG activates endothelial IgG receptor FcγRIIB to promote obesity-induced insulin resistance
Abstract
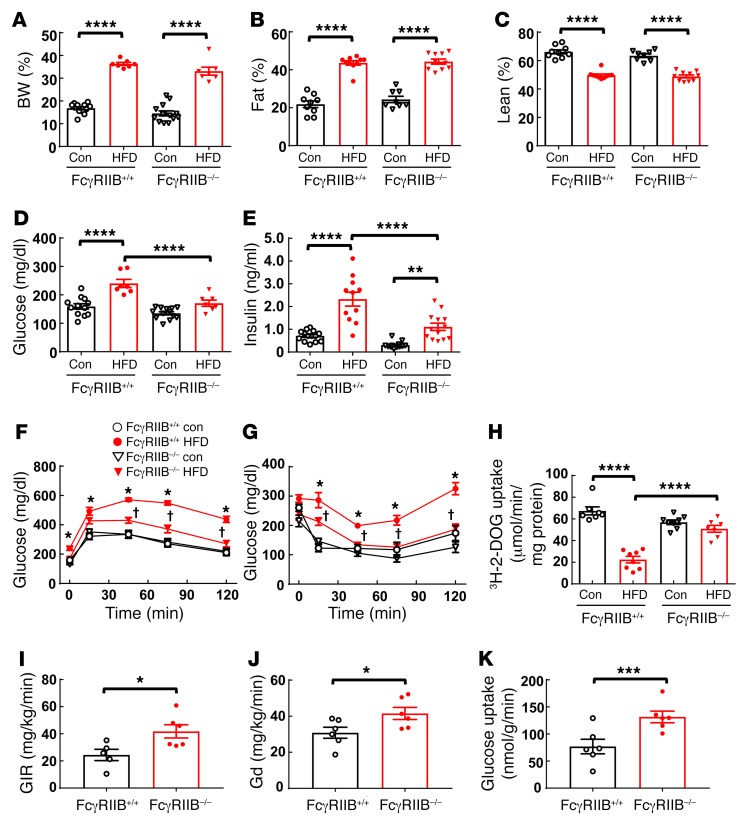
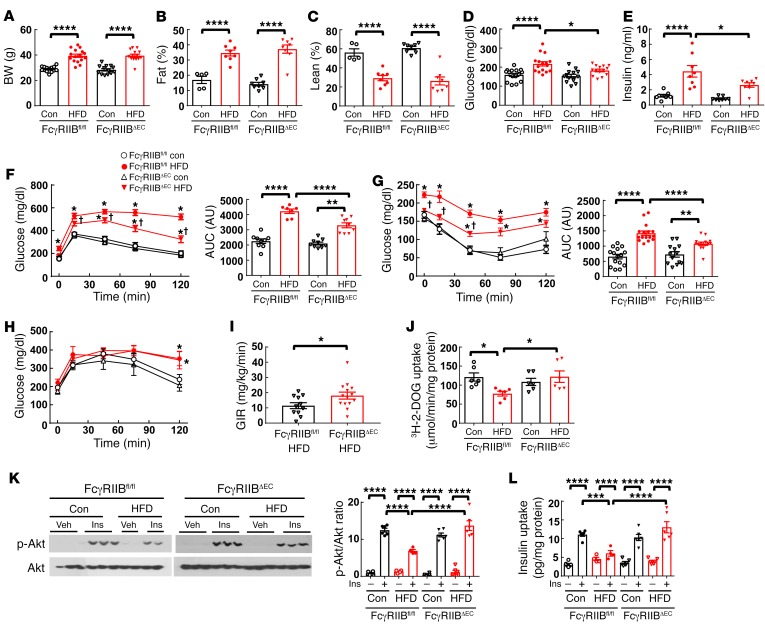
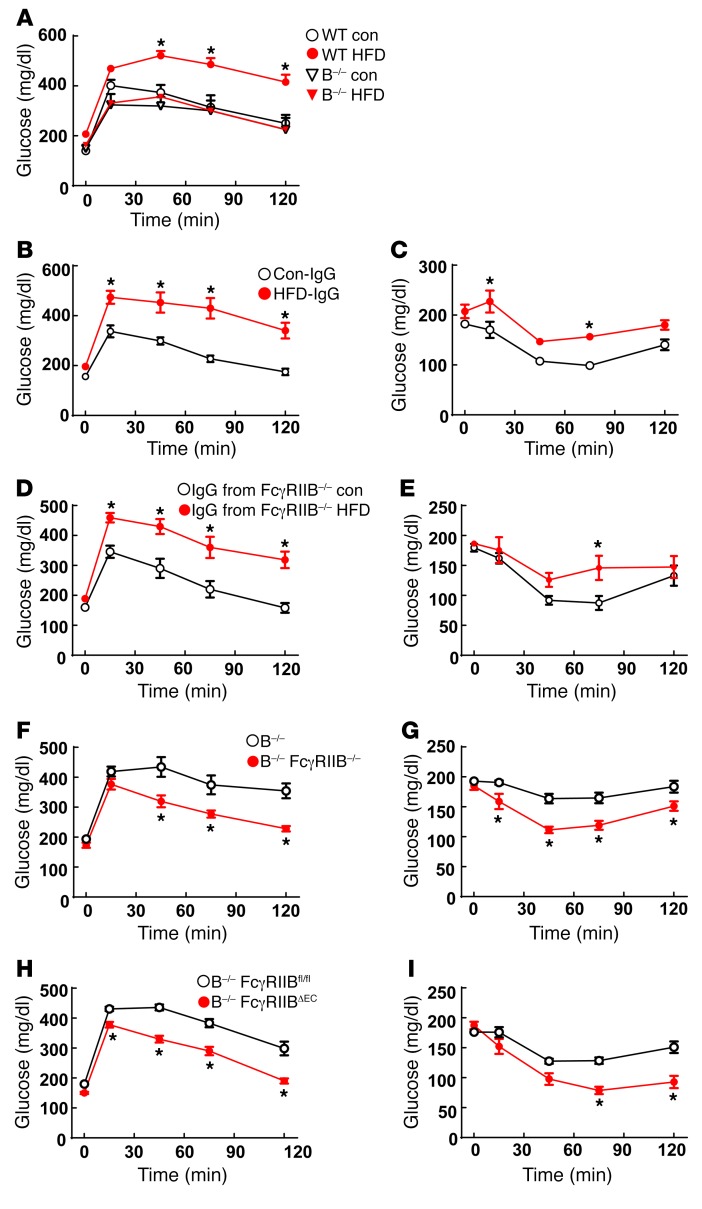
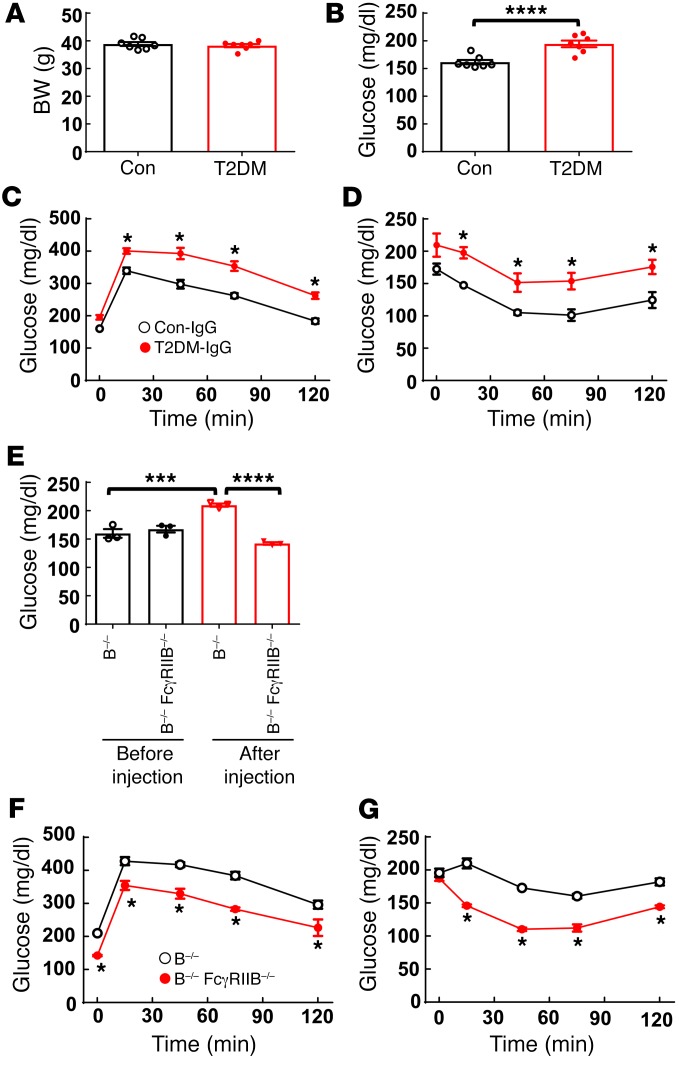
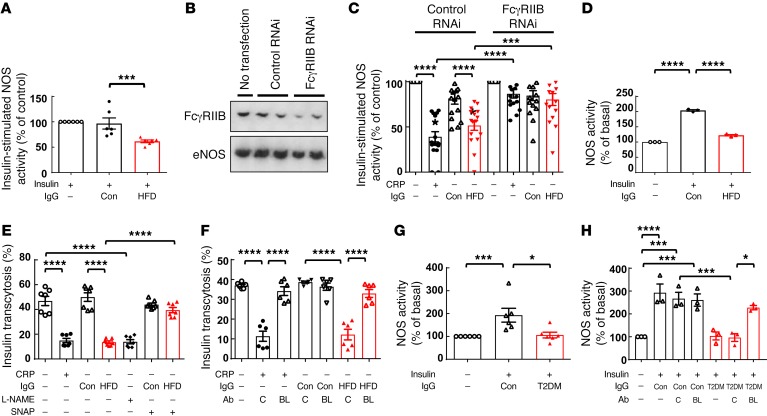
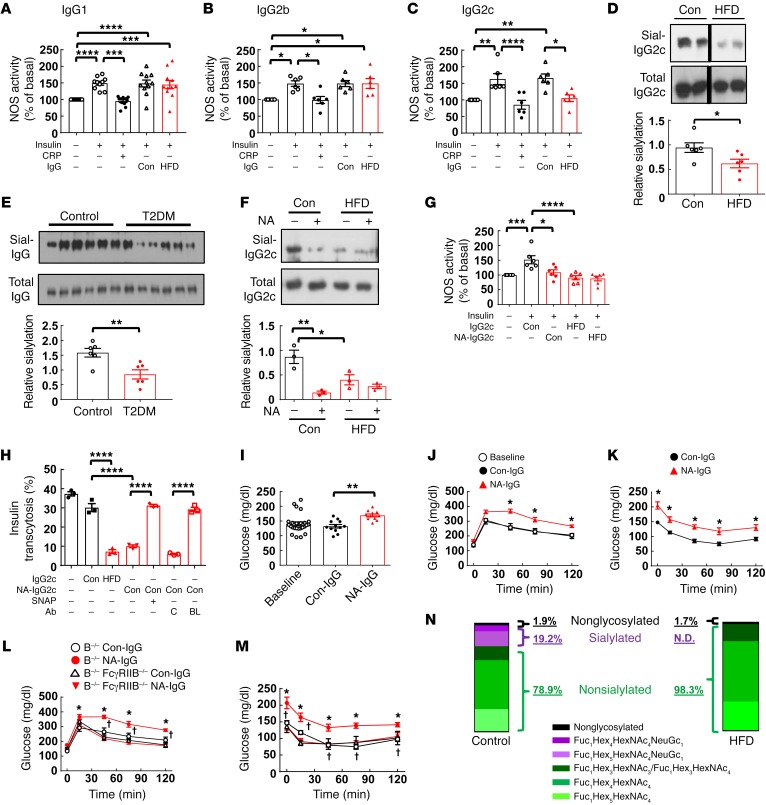
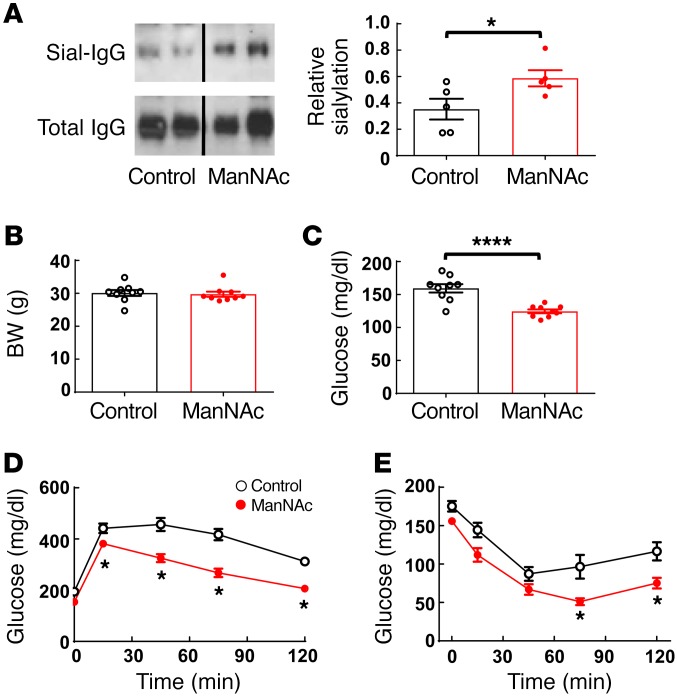
Type 2 diabetes mellitus (T2DM) is a common complication of obesity. Here, we have shown that activation of the IgG receptor FcγRIIB in endothelium by hyposialylated IgG plays an important role in obesity-induced insulin resistance. Despite becoming obese on a high-fat diet (HFD), mice lacking FcγRIIB globally or selectively in endothelium were protected from insulin resistance as a result of the preservation of insulin delivery to skeletal muscle and resulting maintenance of muscle glucose disposal. IgG transfer in IgG-deficient mice implicated IgG as the pathogenetic ligand for endothelial FcγRIIB in obesity-induced insulin resistance. Moreover, IgG transferred from patients with T2DM but not from metabolically healthy subjects caused insulin resistance in IgG-deficient mice via FcγRIIB, indicating that similar processes may be operative in T2DM in humans. Mechanistically, the activation of FcγRIIB by IgG from obese mice impaired endothelial cell insulin transcytosis in culture and in vivo. These effects were attributed to hyposialylation of the Fc glycan, and IgG from T2DM patients was also hyposialylated. In HFD-fed mice, supplementation with the sialic acid precursor N-acetyl-D-mannosamine restored IgG sialylation and preserved insulin sensitivity without affecting weight gain. Thus, IgG sialylation and endothelial FcγRIIB may represent promising therapeutic targets to sever the link between obesity and T2DM.
Keywords: Metabolism; Mouse models; Obesity; Vascular Biology; endothelial cells.
Conflict of interest statement
Figures
Similar articles
-
Supplementation With the Sialic Acid Precursor N-Acetyl-D-Mannosamine Breaks the Link Between Obesity and Hypertension.Circulation. 2019 Dec 10;140(24):2005-2018. doi: 10.1161/CIRCULATIONAHA.119.043490. Epub 2019 Oct 10. Circulation. 2019. PMID: 31597453 Free PMC article.
-
Endothelial Fcγ Receptor IIB Activation Blunts Insulin Delivery to Skeletal Muscle to Cause Insulin Resistance in Mice.Diabetes. 2016 Jul;65(7):1996-2005. doi: 10.2337/db15-1605. Epub 2016 Apr 26. Diabetes. 2016. PMID: 27207525 Free PMC article.
-
IgG receptor FcγRIIB plays a key role in obesity-induced hypertension.Hypertension. 2015 Feb;65(2):456-62. doi: 10.1161/HYPERTENSIONAHA.114.04670. Epub 2014 Nov 3. Hypertension. 2015. PMID: 25368023 Free PMC article.
-
Metabolism and insulin signaling in common metabolic disorders and inherited insulin resistance.Dan Med J. 2014 Jul;61(7):B4890. Dan Med J. 2014. PMID: 25123125 Review.
-
[Achievements in molecular genetics studies of diabetes mellitus].Biomed Khim. 2005 Mar-Apr;51(2):107-17. Biomed Khim. 2005. PMID: 15945346 Review. Russian.
Cited by
-
Leptin receptor deficiency impedes metabolic surgery related-weight loss through inhibition of energy expenditure in db/db mice.Diabetol Metab Syndr. 2024 Feb 1;16(1):33. doi: 10.1186/s13098-024-01270-7. Diabetol Metab Syndr. 2024. PMID: 38302999 Free PMC article.
-
Assessment of Transarterial Chemoembolization Using Super-resolution Ultrasound Imaging and a Rat Model of Hepatocellular Carcinoma.Ultrasound Med Biol. 2023 May;49(5):1318-1326. doi: 10.1016/j.ultrasmedbio.2023.01.021. Epub 2023 Mar 1. Ultrasound Med Biol. 2023. PMID: 36868958 Free PMC article.
-
An Inflammatory Story: Antibodies in Tuberculosis Comorbidities.Front Immunol. 2019 Dec 9;10:2846. doi: 10.3389/fimmu.2019.02846. eCollection 2019. Front Immunol. 2019. PMID: 31921122 Free PMC article. Review.
-
Effects of Estradiol on Immunoglobulin G Glycosylation: Mapping of the Downstream Signaling Mechanism.Front Immunol. 2021 May 25;12:680227. doi: 10.3389/fimmu.2021.680227. eCollection 2021. Front Immunol. 2021. PMID: 34113353 Free PMC article.
-
Generalized mathematical framework for contrast-enhanced ultrasound imaging with pulse inversion spectral deconvolution.Ultrasonics. 2023 Mar;129:106913. doi: 10.1016/j.ultras.2022.106913. Epub 2022 Dec 9. Ultrasonics. 2023. PMID: 36528905 Free PMC article.
References
-
- Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardiometabolic risk factors from 1980 to 2010: a comparative risk assessment. Lancet Diabetes Endocrinol. 2014;2(8):634–647. doi: 10.1016/S2213-8587(14)70102-0. - DOI - PMC - PubMed
-
- Danaei G, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2·7 million participants. Lancet. 2011;378(9785):31–40. doi: 10.1016/S0140-6736(11)60679-X. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
