

Genetic investigation of XPA gene: high frequency of the c.682C>T mutation in Moroccan XP patients with moderate clinical profile
- PMID: 29208038
- PMCID: PMC5718079
- DOI: 10.1186/s13104-017-3042-6
Genetic investigation of XPA gene: high frequency of the c.682C>T mutation in Moroccan XP patients with moderate clinical profile
Abstract
Objective: Xeroderma pigmentosum (XP) is a genetically and clinically heterogeneous disease, associated with an inherited defect in one of eight different genes (XPA to XPG and XPV). In addition to the early onset of the skin manifestations, the XP group A is marked by the presence of a mild to severe neural disorders which appear tardily and worsens with age. In this study, 9 patients with moderate clinical profile belonging to 6 XP families were recruited to determine the XPA mutational spectrum in Morocco, using the direct sequencing of the whole coding region of the XPA gene.
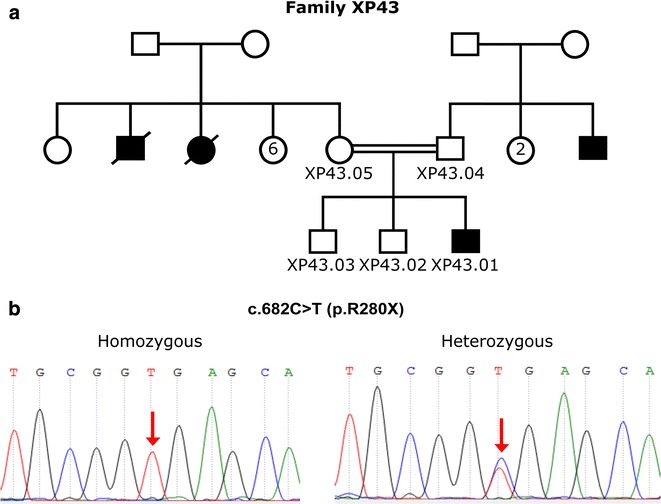
Results: The genetic investigation of the XPA gene showed that 7 from 9 patients were homozygous for the c.682C>T, p.Arg228X mutation, while all their investigated family members were heterozygous. The frequency of this mutation was estimated to be 83.33% (5/6 families) .The molecular analysis of the 5 other exons of the XPA gene, showed that the 2 negative siblings carried no mutation in the XPA gene. This finding suggests that c.682C>T (p.Arg228X) mutation is relatively associated with moderate phenotype in XP group A Moroccan families; this result will also contribute to improve the molecular diagnosis of XP disease in Moroccan patients.
Keywords: Morocco; Mutation; XPA; Xeroderma pigmentosum.
Figures

Similar articles
-
Genetic homogeneity of mutational spectrum of group-A xeroderma pigmentosum in Tunisian patients.Int J Dermatol. 2010 May;49(5):544-8. doi: 10.1111/j.1365-4632.2010.04421.x. Int J Dermatol. 2010. PMID: 20534089
-
Diagnosis of Xeroderma Pigmentosum Groups A and C by Detection of Two Prevalent Mutations in West Algerian Population: A Rapid Genotyping Tool for the Frequent XPC Mutation c.1643_1644delTG.Biomed Res Int. 2016;2016:2180946. doi: 10.1155/2016/2180946. Epub 2016 Jun 20. Biomed Res Int. 2016. PMID: 27413738 Free PMC article.
-
Mutational spectrum of Xeroderma pigmentosum group A in Egyptian patients.Gene. 2014 Jan 1;533(1):52-6. doi: 10.1016/j.gene.2013.09.125. Epub 2013 Oct 14. Gene. 2014. PMID: 24135642
-
A novel mutation in the XPA gene associated with unusually mild clinical features in a patient who developed a spindle cell melanoma.Br J Dermatol. 2006 Jul;155(1):81-8. doi: 10.1111/j.1365-2133.2006.07272.x. Br J Dermatol. 2006. PMID: 16792756 Review.
-
Molecular genetics of Xeroderma pigmentosum variant.Exp Dermatol. 2003 Oct;12(5):529-36. doi: 10.1034/j.1600-0625.2003.00124.x. Exp Dermatol. 2003. PMID: 14705792 Review.
Cited by
-
Case Report: Identification of a Heterozygous XPA c.553C>T Mutation Causing Neurological Impairment in a Case of Xeroderma Pigmentosum Complementation Group A.Front Genet. 2021 Aug 16;12:717361. doi: 10.3389/fgene.2021.717361. eCollection 2021. Front Genet. 2021. PMID: 34484303 Free PMC article.
-
Identification of a ERCC5 c.2333T>C (L778P) Variant in Two Tunisian Siblings With Mild Xeroderma Pigmentosum Phenotype.Front Genet. 2019 Feb 14;10:111. doi: 10.3389/fgene.2019.00111. eCollection 2019. Front Genet. 2019. PMID: 30838033 Free PMC article.
-
Different germline variants in the XPA gene are associated with severe, intermediate, or mild neurodegeneration in xeroderma pigmentosum patients.PLoS Genet. 2024 Dec 2;20(12):e1011265. doi: 10.1371/journal.pgen.1011265. eCollection 2024 Dec. PLoS Genet. 2024. PMID: 39621777 Free PMC article.
References
-
- Butt FMA, Moshi JR, Owibingire S, Chindia ML. Xeroderma pigmentosum: a review and case series. J Cranio-Maxillo-fac Surg Off Publ Eur Assoc Cranio-Maxillo-fac Surg. 2010;38(7):534–537. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
