

Dual Molecular Mechanisms Govern Escape at Immunodominant HLA A2-Restricted HIV Epitope
- PMID: 29209312
- PMCID: PMC5701626
- DOI: 10.3389/fimmu.2017.01503
Dual Molecular Mechanisms Govern Escape at Immunodominant HLA A2-Restricted HIV Epitope
Abstract
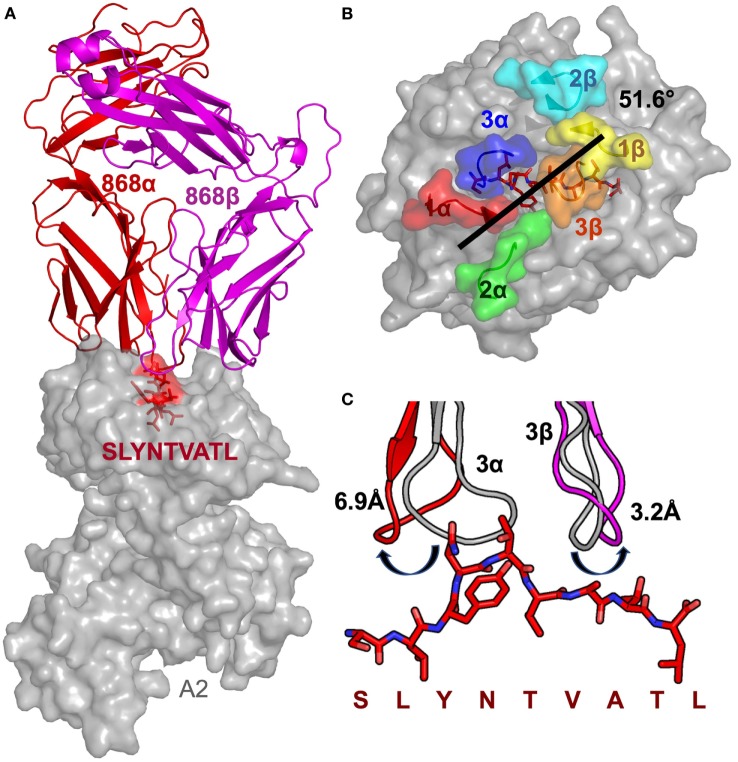
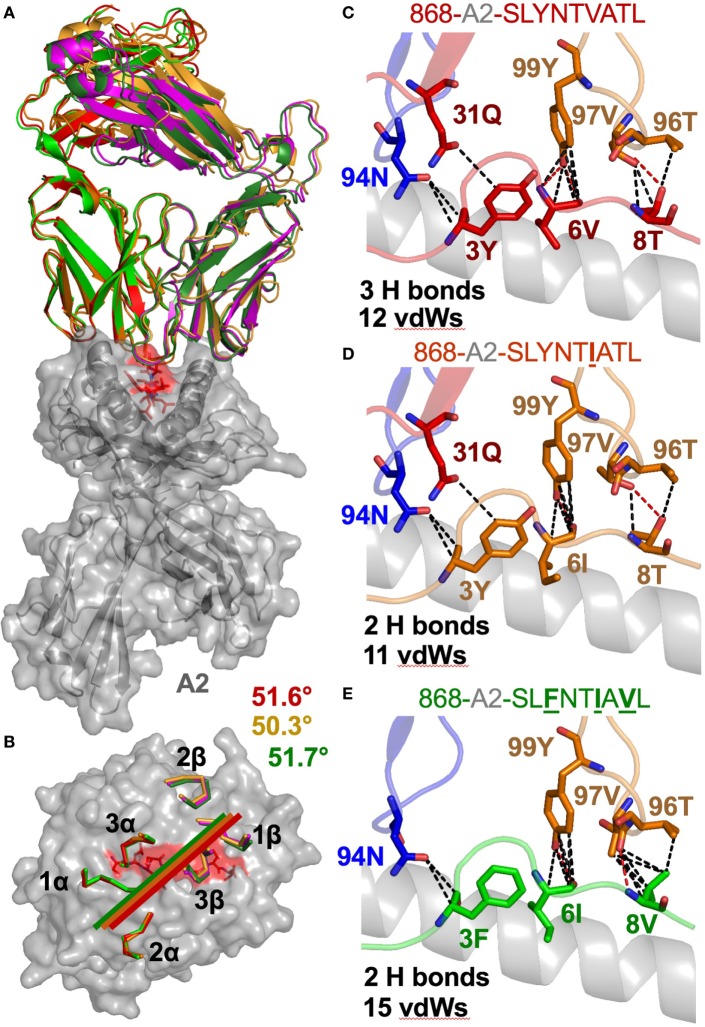
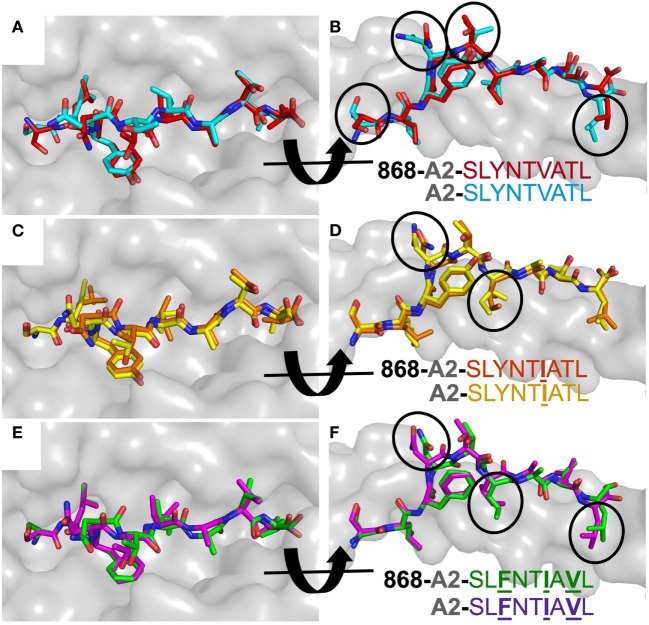
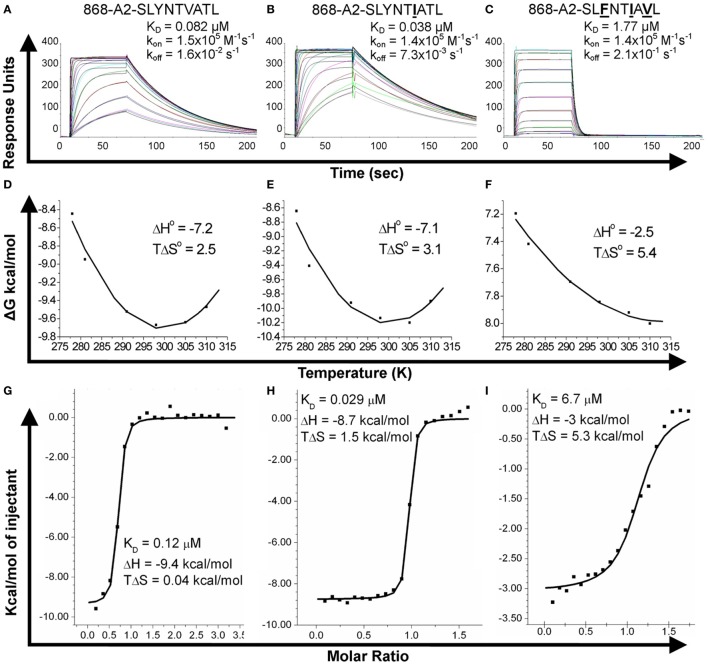
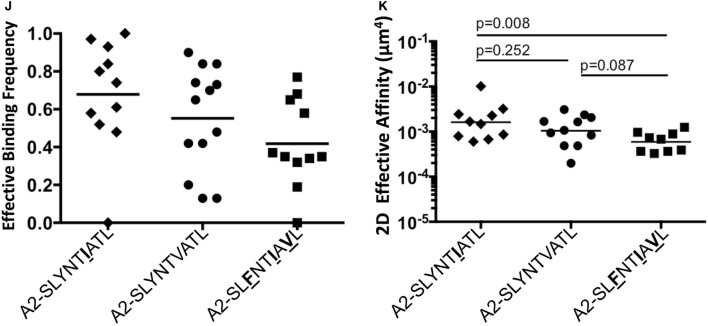
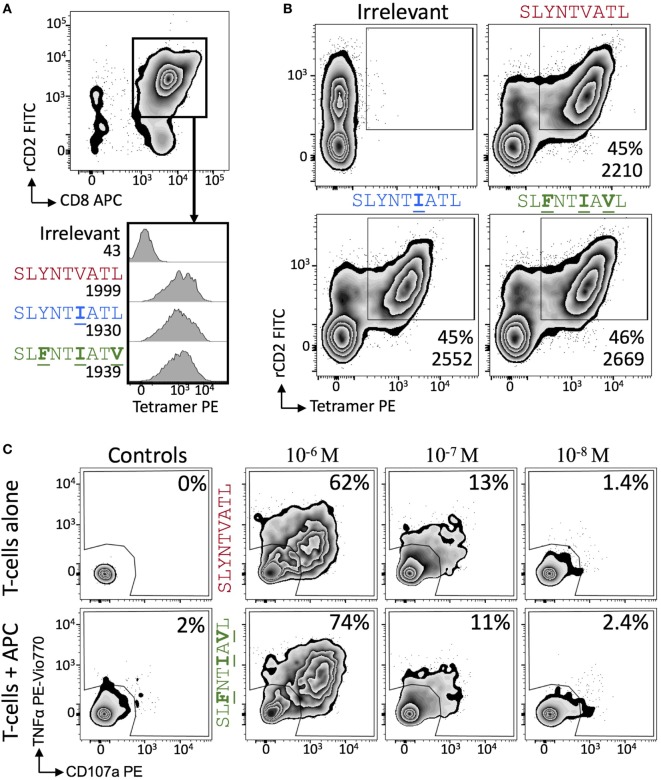
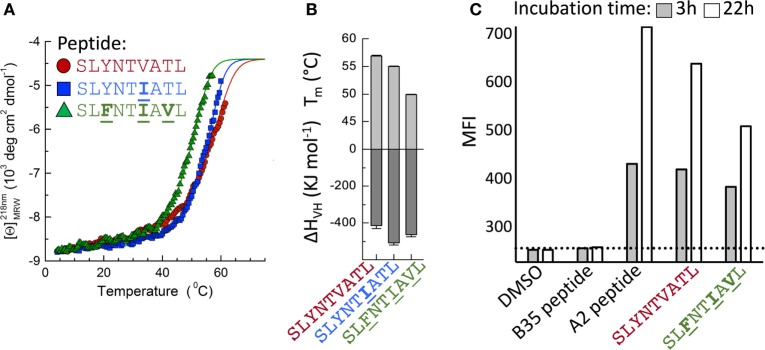
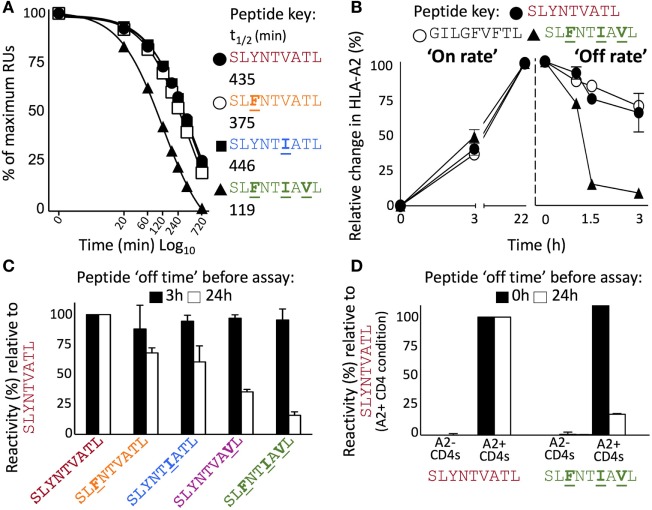
Serial accumulation of mutations to fixation in the SLYNTVATL (SL9) immunodominant, HIV p17 Gag-derived, HLA A2-restricted cytotoxic T lymphocyte epitope produce the SLFNTIAVL triple mutant "ultimate" escape variant. These mutations in solvent-exposed residues are believed to interfere with TCR recognition, although confirmation has awaited structural verification. Here, we solved a TCR co-complex structure with SL9 and the triple escape mutant to determine the mechanism of immune escape in this eminent system. We show that, in contrast to prevailing hypotheses, the main TCR contact residue is 4N and the dominant mechanism of escape is not via lack of TCR engagement. Instead, mutation of solvent-exposed residues in the peptide destabilise the peptide-HLA and reduce peptide density at the cell surface. These results highlight the extraordinary lengths that HIV employs to evade detection by high-affinity TCRs with a broad peptide-binding footprint and necessitate re-evaluation of this exemplar model of HIV TCR escape.
Keywords: HIV; MHC; T-cell; T-cell receptor; immune escape.
Figures

Similar articles
-
Recognition patterns of HLA-A2-restricted human immunodeficiency virus-1-specific cytotoxic T-lymphocytes in a cohort of HIV-1-infected individuals.Viral Immunol. 2005;18(4):627-36. doi: 10.1089/vim.2005.18.627. Viral Immunol. 2005. PMID: 16359229
-
Structural basis for degenerate recognition of natural HIV peptide variants by cytotoxic lymphocytes.J Biol Chem. 2006 Jul 21;281(29):20205-12. doi: 10.1074/jbc.M601934200. Epub 2006 May 15. J Biol Chem. 2006. PMID: 16702212
-
Efficient processing of the immunodominant, HLA-A*0201-restricted human immunodeficiency virus type 1 cytotoxic T-lymphocyte epitope despite multiple variations in the epitope flanking sequences.J Virol. 1999 Dec;73(12):10191-8. doi: 10.1128/JVI.73.12.10191-10198.1999. J Virol. 1999. PMID: 10559335 Free PMC article.
-
Mother-to-child transmission of HIV infection and CTL escape through HLA-A2-SLYNTVATL epitope sequence variation.Immunol Lett. 2001 Nov 1;79(1-2):109-16. doi: 10.1016/s0165-2478(01)00272-3. Immunol Lett. 2001. PMID: 11595297
-
The HIV-1 HLA-A2-SLYNTVATL is a help-independent CTL epitope.J Immunol. 2004 May 1;172(9):5249-61. doi: 10.4049/jimmunol.172.9.5249. J Immunol. 2004. PMID: 15100263
Cited by
-
Nonstimulatory peptide-MHC enhances human T-cell antigen-specific responses by amplifying proximal TCR signaling.Nat Commun. 2018 Jul 13;9(1):2716. doi: 10.1038/s41467-018-05288-0. Nat Commun. 2018. PMID: 30006605 Free PMC article.
-
Molecular Basis of a Dominant SARS-CoV-2 Spike-Derived Epitope Presented by HLA-A*02:01 Recognised by a Public TCR.Cells. 2021 Oct 3;10(10):2646. doi: 10.3390/cells10102646. Cells. 2021. PMID: 34685626 Free PMC article.
-
SARS-CoV-2 infection establishes a stable and age-independent CD8+ T cell response against a dominant nucleocapsid epitope using restricted T cell receptors.Nat Commun. 2023 Oct 23;14(1):6725. doi: 10.1038/s41467-023-42430-z. Nat Commun. 2023. PMID: 37872153 Free PMC article.
-
Characterization of amino acid residues of T-cell receptors interacting with HLA-A*02-restricted antigen peptides.Ann Transl Med. 2021 Mar;9(6):495. doi: 10.21037/atm-21-835. Ann Transl Med. 2021. PMID: 33850892 Free PMC article.
-
Allosteric activation of T cell antigen receptor signaling by quaternary structure relaxation.Cell Rep. 2021 Jul 13;36(2):109375. doi: 10.1016/j.celrep.2021.109375. Cell Rep. 2021. PMID: 34260912 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials