

CAR T-cell immunotherapy of MET-expressing malignant mesothelioma
- PMID: 29209570
- PMCID: PMC5706532
- DOI: 10.1080/2162402X.2017.1363137
CAR T-cell immunotherapy of MET-expressing malignant mesothelioma
Abstract
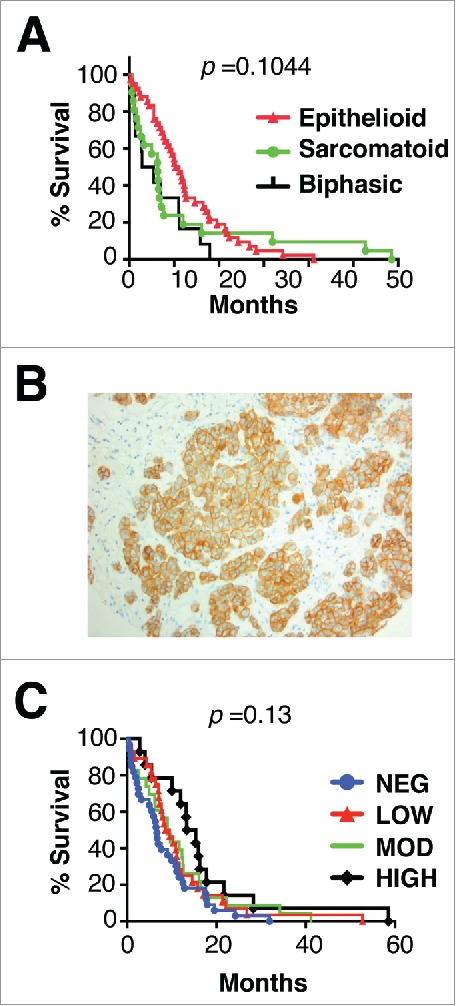
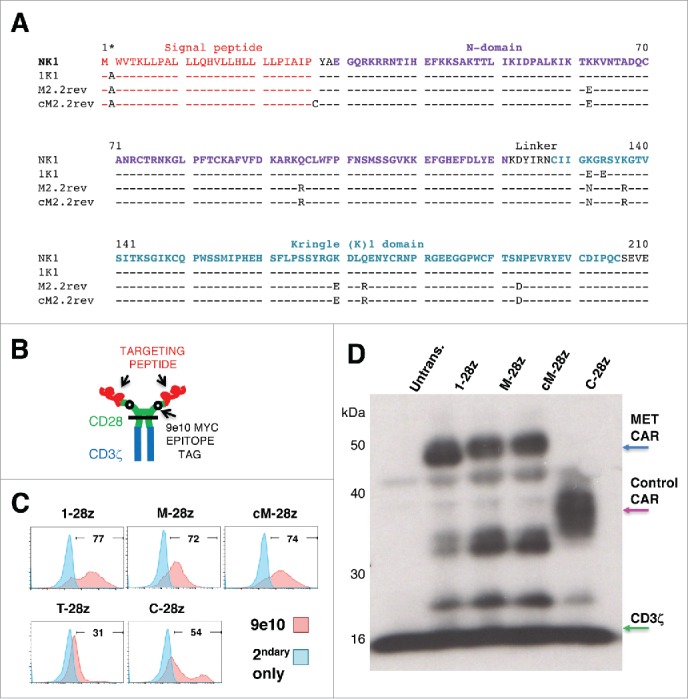
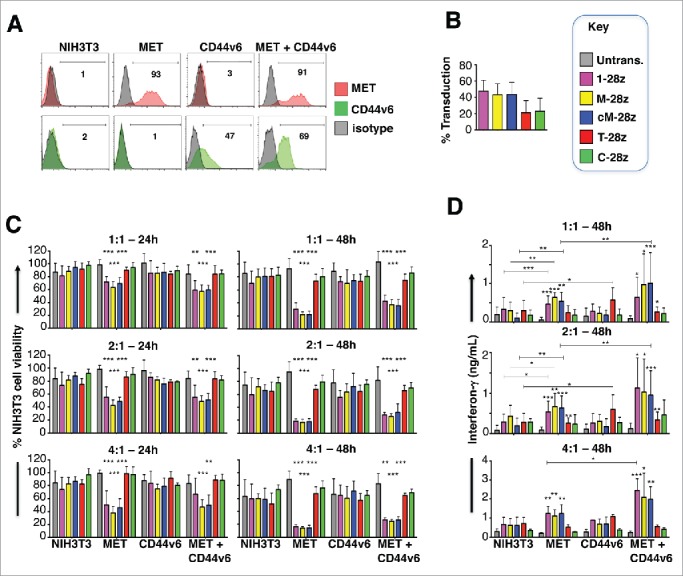
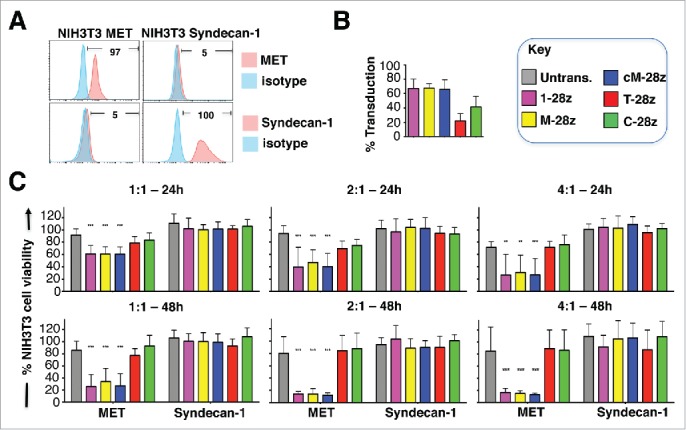
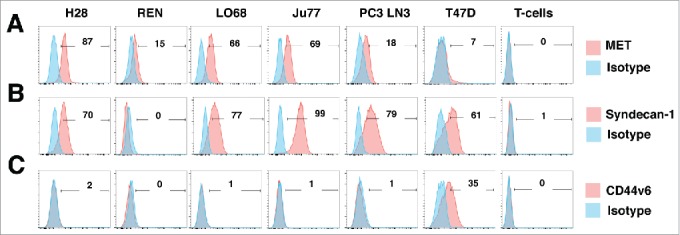
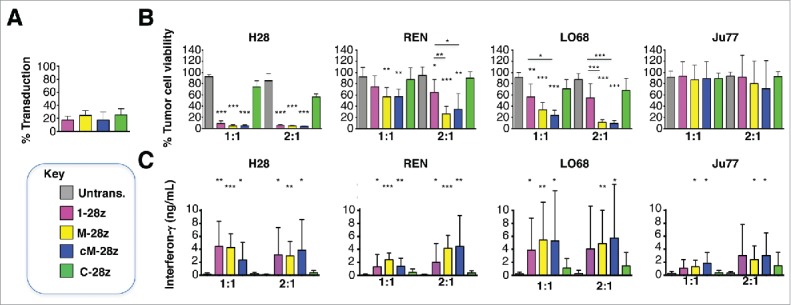
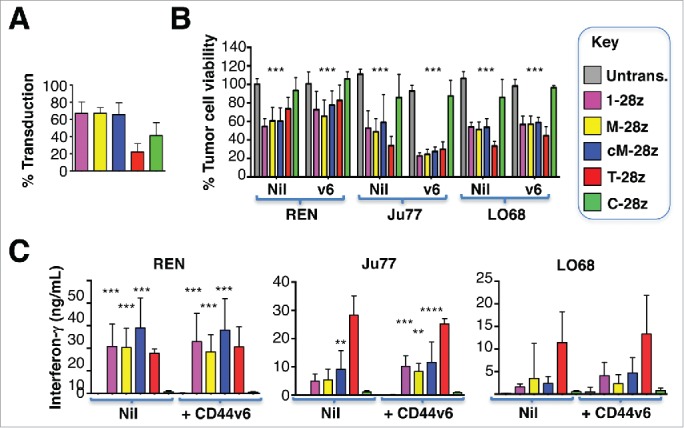
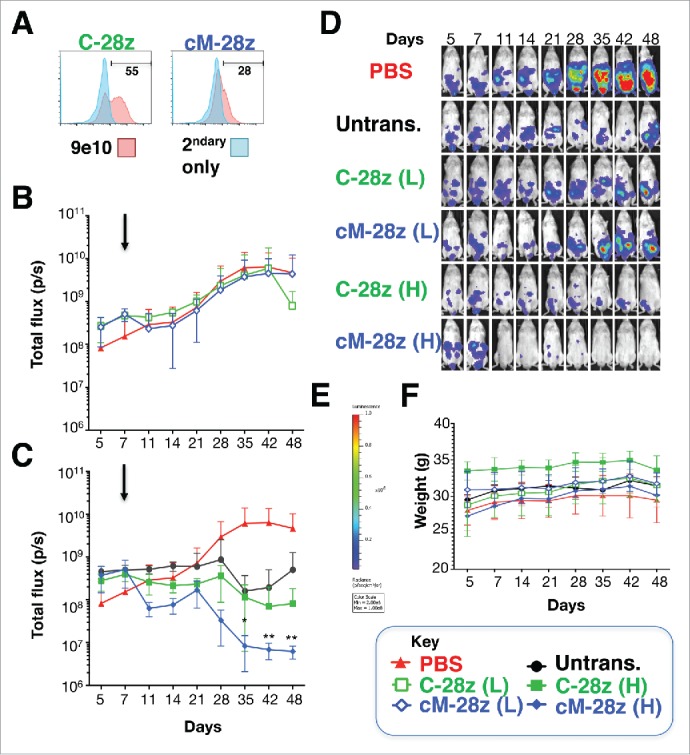
Mesothelioma is an incurable cancer for which effective therapies are required. Aberrant MET expression is prevalent in mesothelioma, although targeting using small molecule-based therapeutics has proven disappointing. Chimeric antigen receptors (CARs) couple the HLA-independent binding of a cell surface target to the delivery of a tailored T-cell activating signal. Here, we evaluated the anti-tumor activity of MET re-targeted CAR T-cells against mesothelioma. Using immunohistochemistry, MET was detected in 67% of malignant pleural mesotheliomas, most frequently of epithelioid or biphasic subtype. The presence of MET did not influence patient survival. Candidate MET-specific CARs were engineered in which a CD28+CD3ζ endodomain was fused to one of 3 peptides derived from the N and K1 domains of hepatocyte growth factor (HGF), which represents the minimum MET binding element present in this growth factor. Using an NIH3T3-based artificial antigen-presenting cell system, we found that all 3 candidate CARs demonstrated high specificity for MET. By contrast, these CARs did not mediate T-cell activation upon engagement of other HGF binding partners, namely CD44v6 or heparan sulfate proteoglycans, including Syndecan-1. NK1-targeted CARs demonstrated broadly similar in vitro potency, indicated by destruction of MET-expressing mesothelioma cell lines, accompanied by cytokine release. In vivo anti-tumor activity was demonstrated following intraperitoneal delivery to mice with an established mesothelioma xenograft. Progressive tumor regression occurred without weight loss or other clinical indicators of toxicity. These data confirm the frequent expression of MET in malignant pleural mesothelioma and demonstrate that this can be targeted effectively and safely using a CAR T-cell immunotherapeutic strategy.
Keywords: Immunotherapy; MET; NK1; cancer; chimeric antigen receptor; mesothelioma.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous