

Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators
- PMID: 29211708
- PMCID: PMC5774979
- DOI: 10.1038/nature25179
Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators
Abstract
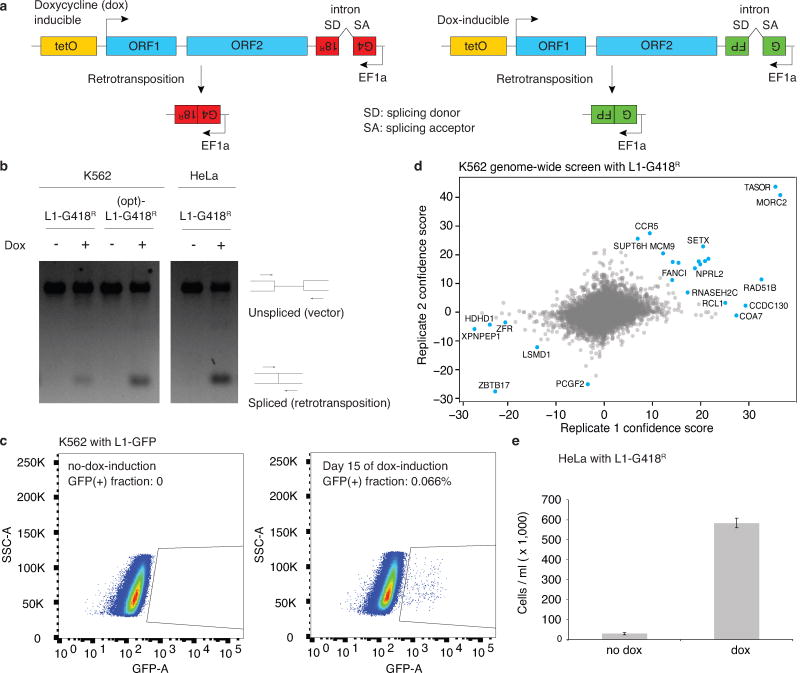
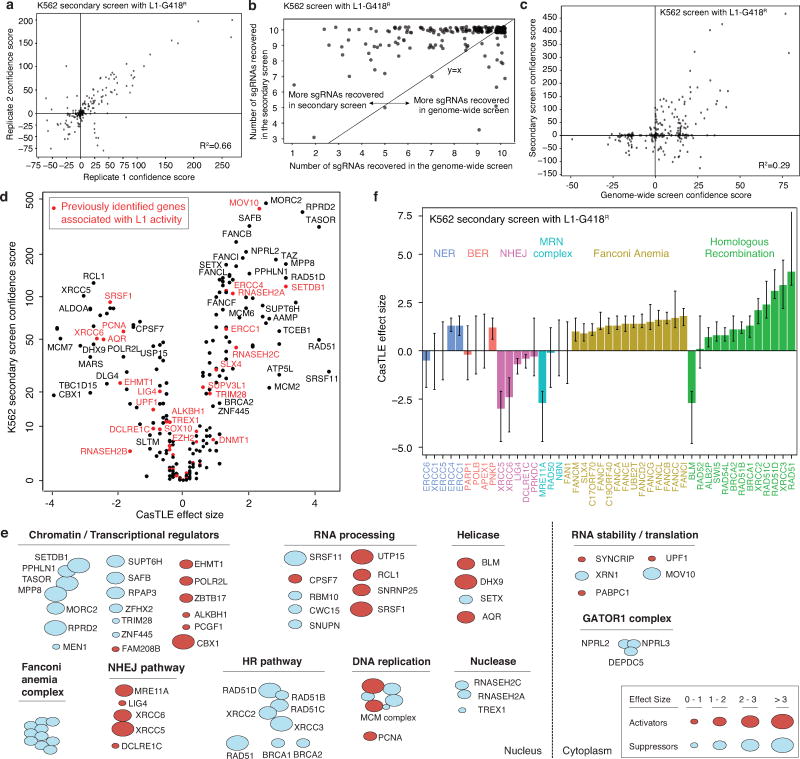
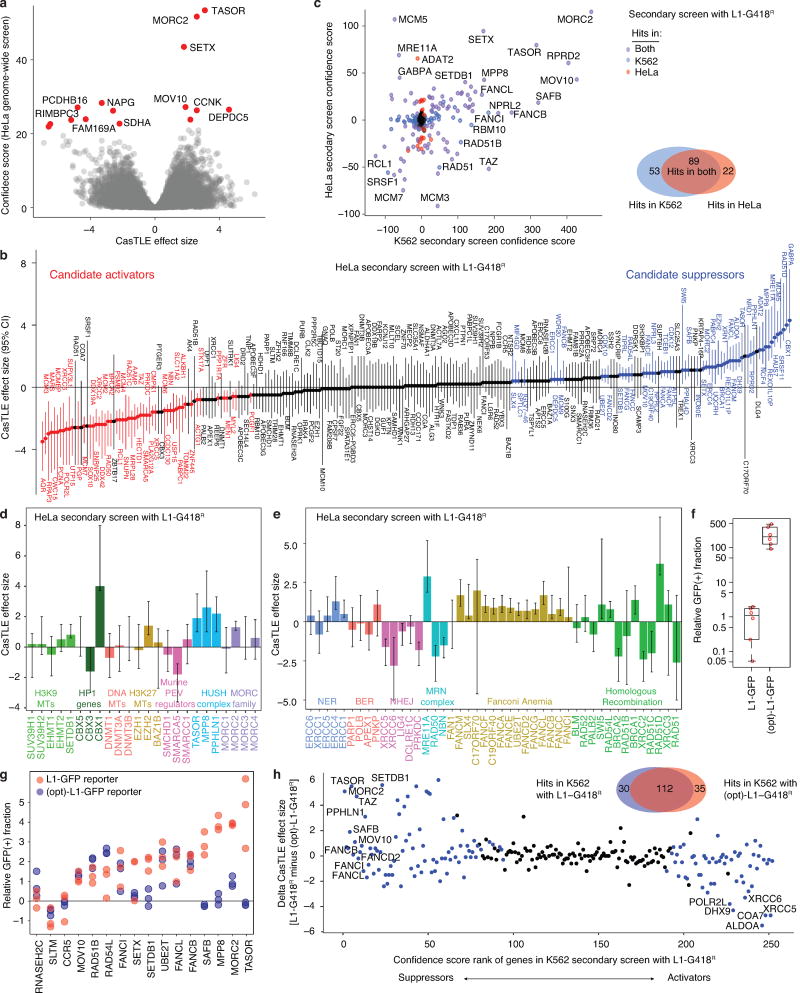
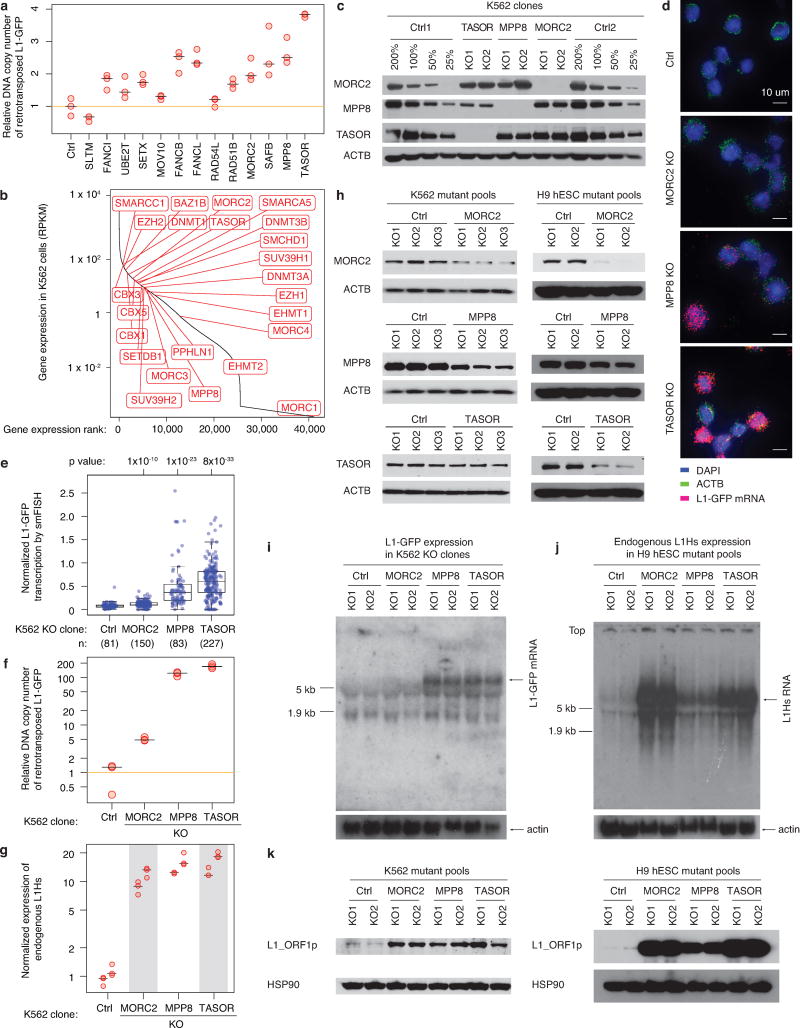
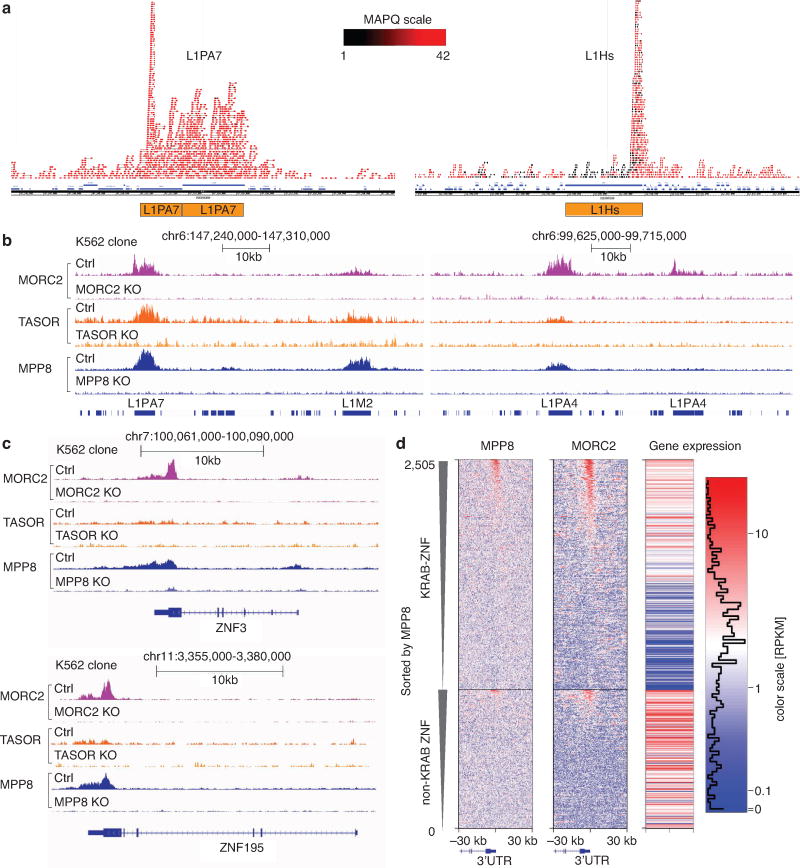
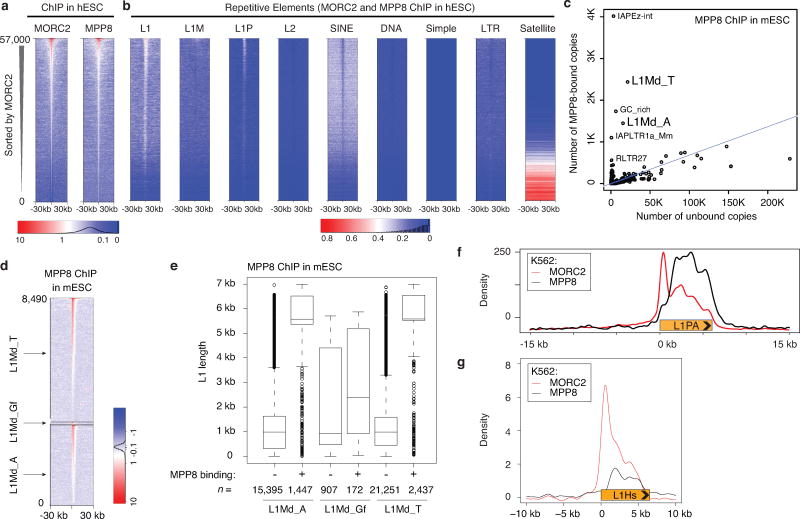
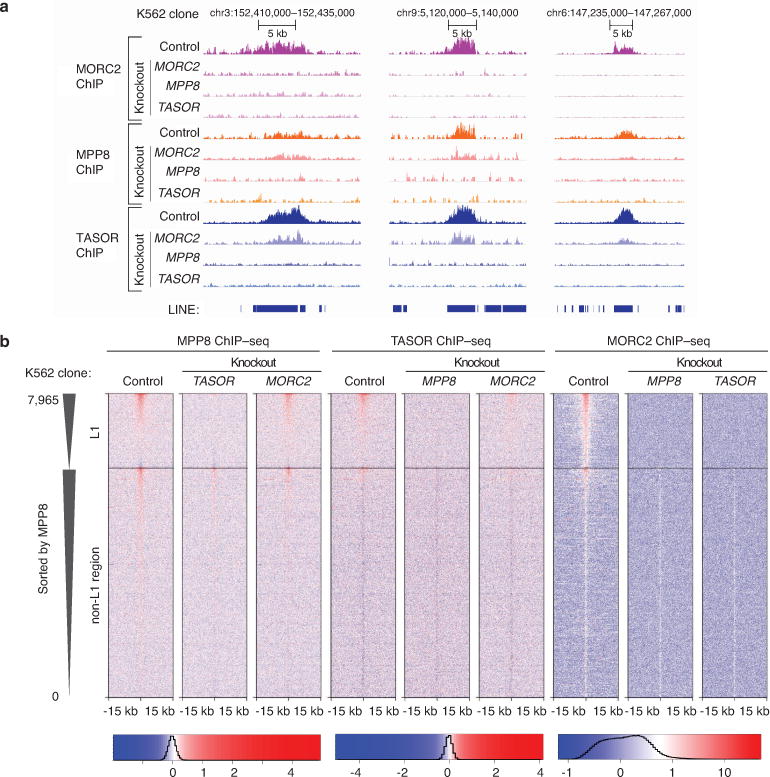
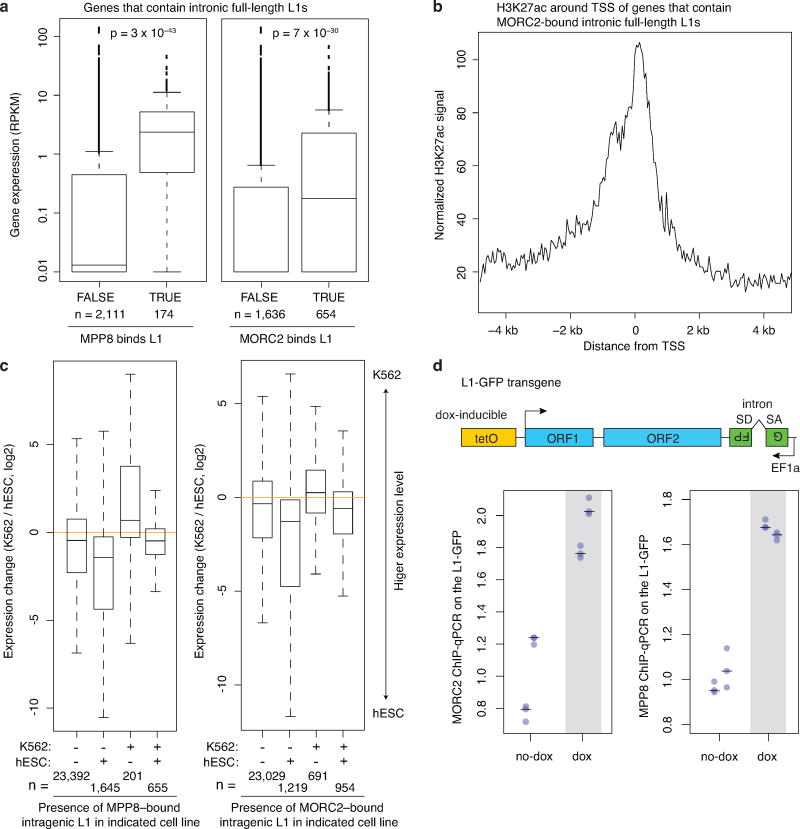
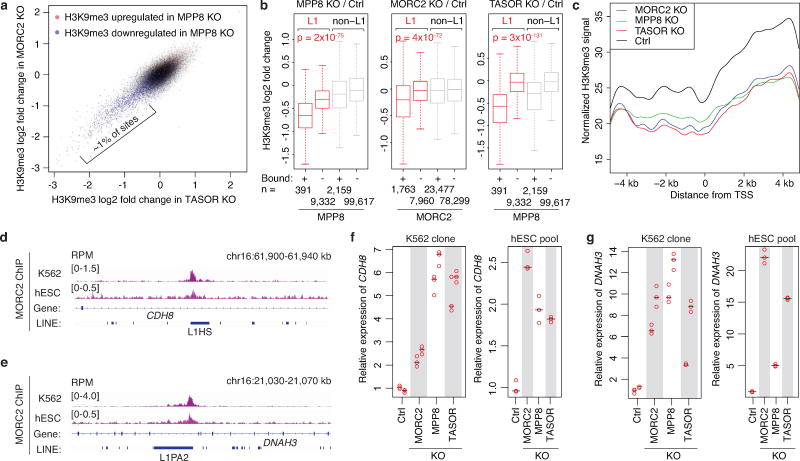
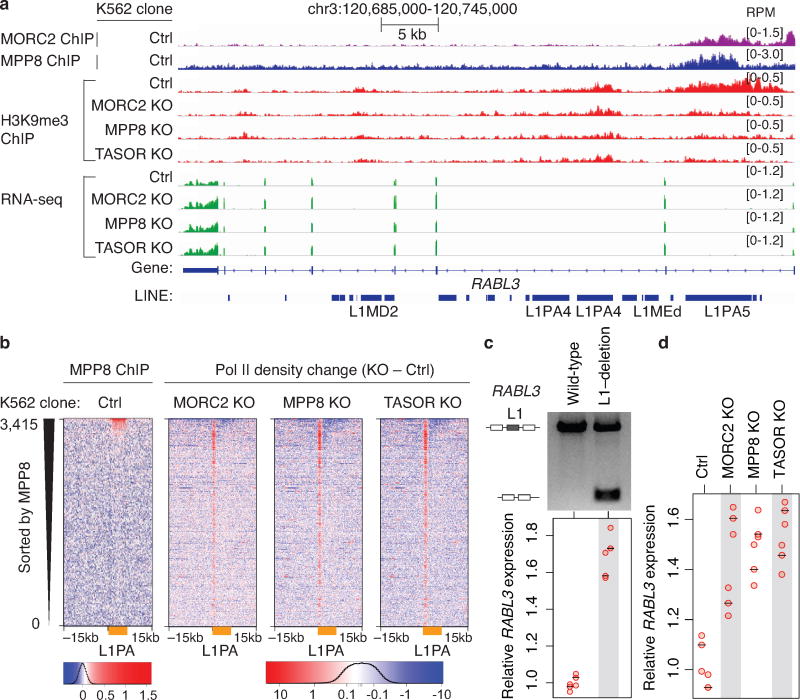
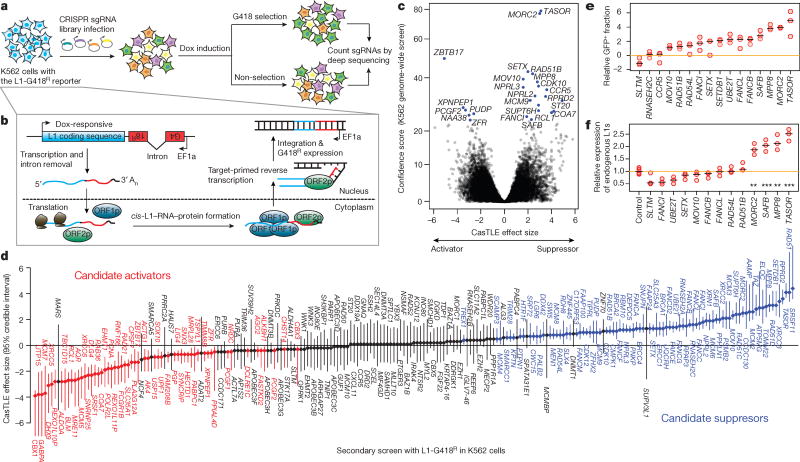
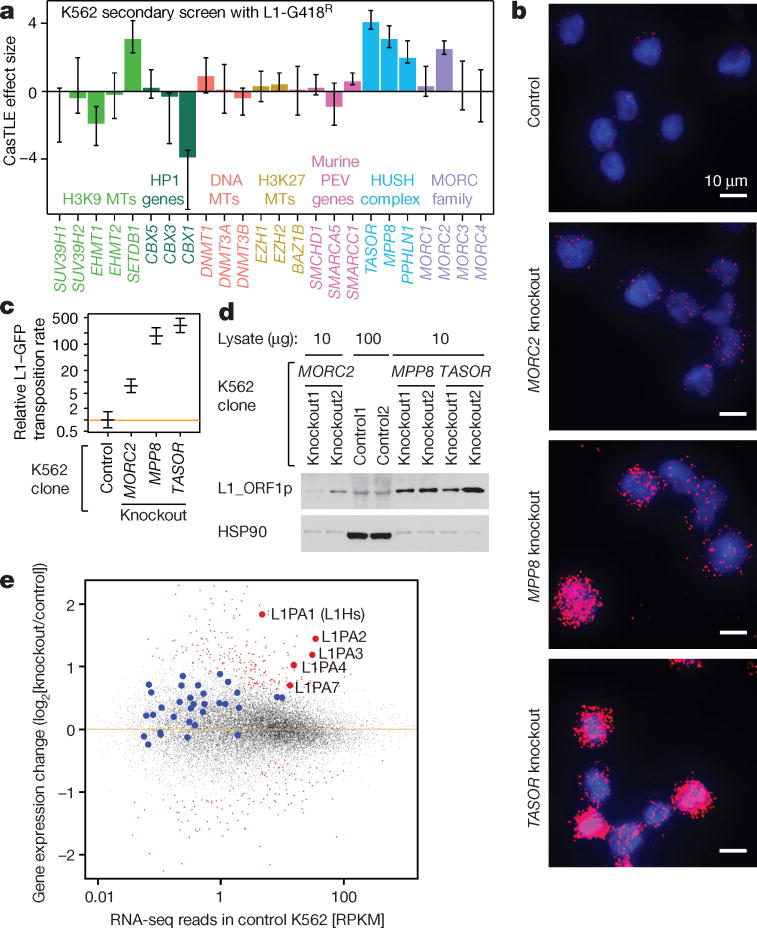
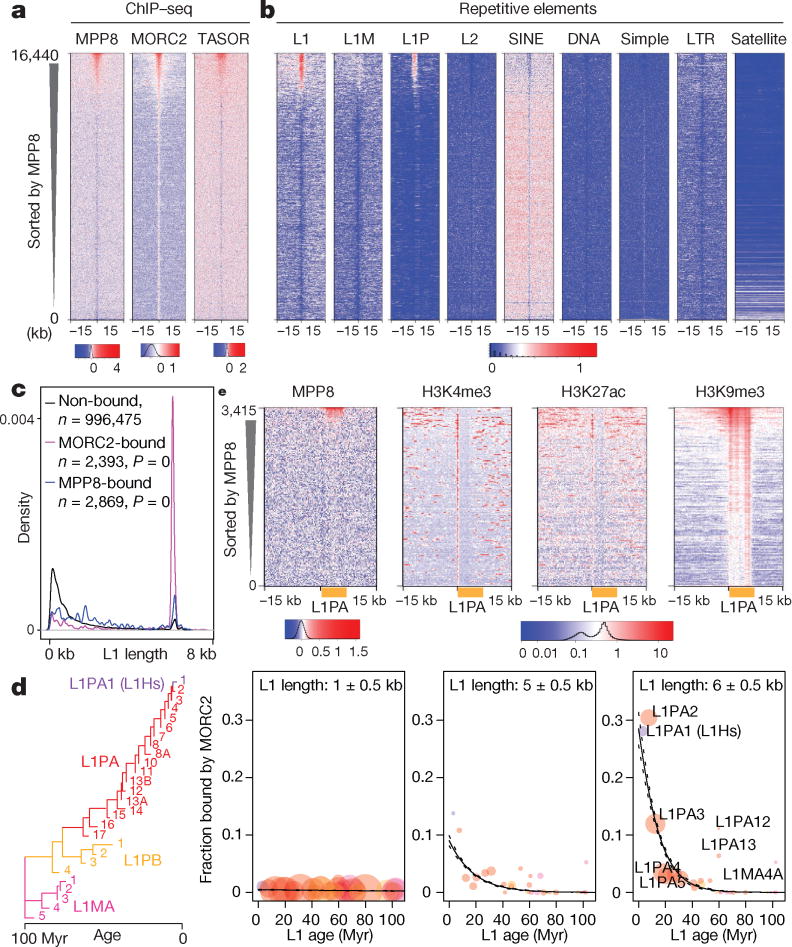
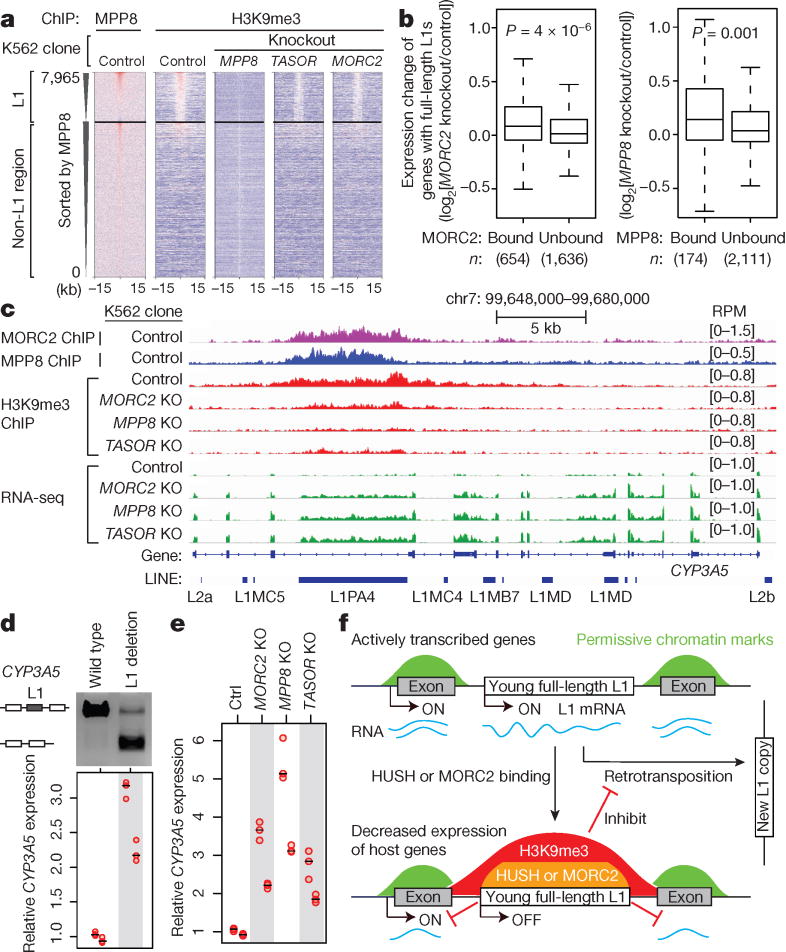
Transposable elements, also known as transposons, are now recognized not only as parasitic DNA, the spread of which in the genome must be controlled by the host, but also as major players in genome evolution and regulation. Long interspersed element-1 (LINE-1, also known as L1), the only currently autonomous mobile transposon in humans, occupies 17% of the genome and generates inter- and intra-individual genetic variation, in some cases resulting in disease. However, how L1 activity is controlled and the function of L1s in host gene regulation are not completely understood. Here we use CRISPR-Cas9 screening strategies in two distinct human cell lines to provide a genome-wide survey of genes involved in the control of L1 retrotransposition. We identify functionally diverse genes that either promote or restrict L1 retrotransposition. These genes, which are often associated with human diseases, control the L1 life cycle at the transcriptional or the post-transcriptional level in a manner that can depend on the endogenous L1 nucleotide sequence, underscoring the complexity of L1 regulation. We further investigate the restriction of L1 by the protein MORC2 and by the human silencing hub (HUSH) complex subunits MPP8 and TASOR. HUSH and MORC2 can selectively bind evolutionarily young, full-length L1s located within transcriptionally permissive euchromatic environments, and promote deposition of histone H3 Lys9 trimethylation (H3K9me3) for transcriptional silencing. Notably, these silencing events often occur within introns of transcriptionally active genes, and lead to the downregulation of host gene expression in a HUSH-, MORC2-, and L1-dependent manner. Together, these results provide a rich resource for studies of L1 retrotransposition, elucidate a novel L1 restriction pathway and illustrate how epigenetic silencing of transposable elements rewires host gene expression programs.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
